
Abstract
Background
Cyanosis is usually associated with serious conditions requiring urgent treatment in the neonatal intensive care unit (NICU). Hemoglobin M (Hb M) disease is one type of congenital methemoglobinemia characterized by cyanosis. Among these variants, α-globin chain mutations such as Hb M Boston present cyanosis from birth while other variants usually manifest later in life.
Case presentation
We report a case of a male newborn with cyanosis apparent since birth. Surprisingly, his respiratory and hemodynamic status including normal arterial blood oxygen saturation was stable, but oxygen saturation on pulse oximetry did not increase after 100% supplemental oxygen was started. In addition to routine pulmonary and cardiologic evaluation, further evaluation for dyshemoglobin was conducted; α2-globin gene sequencing showed a single-point variant causing Hb M Boston. Methemoglobin (MetHb) level estimated by co-oximetry was normal. After a 14-day stay in the NICU, the patient remained respiratory and hemodynamically stable without supplemental oxygen except for cyanosis.
Conclusions
Hb M disease is a benign disease and does not require any treatment whereas acquired methemoglobinemia is a potentially fatal condition. Neonatologists should be aware that low oxygenation status on pulse oximetry in the face of normal arterial blood saturation values might indicate the possibility of Hb M disease in early neonatal cyanosis, irrespective of MetHb value.
Similar content being viewed by others

Background
Hemoglobin M (Hb M) is one of the causes of inherited methemoglobinemia. Methemoglobin (MetHb) refers to the oxidized form of hemoglobin. Hb M caused by a mutation in α-, β-, or γ-globin can lead to spontaneous oxidation of the ferrous ion in the heme and cannot transport or release oxygen in tissues. Hb M disease causes cyanosis that is unresponsive to oxygen therapy [1, 2]. Some variants might need to be considered in the differential diagnosis of central cyanosis in newborns because they cause cyanosis from birth. Hemoglobin variants by a genetic mutation are different from natural MetHb in the absorption spectrum. Then, co-oximetric measurement may be inaccurate in cases of Hb M [3].
Unlike other types of methemoglobinemia, Hb M disease is a benign disease and does not require any treatment although it reveals a history of lifelong cyanosis. Clinical suspicion and early diagnosis could avoid unnecessary investigations and invasive management for patients and alleviate concern for their family [4, 5]. In this study, we present a male newborn with Hb M Boston and review the literature for Hb M disease since 1961.
Case presentation
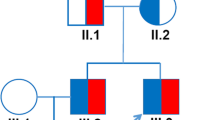
A male newborn was referred to the neonatal intensive care unit with cyanosis from birth. After applying supplemental oxygen, pre- and postductal saturation was 80%. Tracheal intubation and positive pressure ventilation with high inspired oxygen concentration did not improve the low oxygen saturation (SO2) level. The infant weighed 3400 g and was delivered uneventfully at 38 + 6 weeks from a 32-year-old mother. He did not appear to have any signs of respiratory difficulties except for cyanosis. When SO2 was 77% according to pulse oximetry, the value of partial pressure of oxygen (PaO2) on arterial blood gas analysis was 114 mmHg. His complete blood count and C-reactive protein were normal. After ruling out respiratory and cardiac causes based on chest radiographic and echocardiographic studies, further evaluation for hemoglobin derivatives incapable of binding oxygen was conducted. MetHb level estimated by co-oximetry was found to be normal. In addition, hemoglobin electrophoresis at alkaline pH on agarose gel showed normal age profiles with 82.7% Hb F, 17.2% Hb A1, and 0% Hb S. Exon sequencing was conducted for the α-globin chain of hemoglobin because of the cyanosis at birth. We found a c.175C > T point variant of HBA2 exon 2, which causes a change of the 59th amino acid from histidine to tyrosine (Fig. 1a). This single-point variant causes Hb M Boston. The genetic evaluation of HBA2 in his parents was normal for the above mutation (Fig. 1b); this appeared to be a de novo mutation. Gene sequencing of erythroid Krüppel-like factor for thalassemia and CYB5R3 for methemoglobinemia types 1 and 2 was negative.

a The mutation underlying heterozygous hemoglobin M Boston [HBA2:c.175C > T, p.59His > Tyr] was detected through sequencing only in the proband. b It was not present in his parents. The analysis demonstrated a de novo mutation occurrence
At discharge, the patient remained respiratory and hemodynamically stable without supplemental oxygen except for cyanosis. At 2 years of age, his weight was 16.5 kg and height was 95.5 cm, placing him in the 99th percentile according to national growth charts for children; however, he remained slightly cyanotic at 83% saturation without supplemental oxygen.
Discussion
Since chemical characterizations of Hb M were first reported in the late 1950s and early 1960s by Gerald [6, 7], the rare globin chain variants have been investigated by chromatography, electrophoresis, and gene sequencing [1, 2, 5].
This is the first de novo case of Hb M Boston reported from South Korea [8]. Like other types of Hb M disease, Hb M Boston has an autosomal dominant inheritance pattern [5]. Although de novo mutations are not rare events in human genetic diseases, relatively few have been recorded for Hb M variants and α-chain mutants are rarer than β-chain mutants [2, 5, 9,10,11,12]. This report clearly describes a de novo mutation that was present in the proband by gene sequencing of HBA2. Hb M should be considered in the differential diagnosis of cyanosis in the newborn period, even if no familial cases are detected. Table 1 lists the cases of hemoglobin M disesase with neonatal cynosis presenting immediately after birth.
Hemoglobin is constantly being oxidized; however, natural reducing systems such as erythrocyte MetHb reductase maintain the natural MetHb level under 2% [2, 5]. Hb M variants caused by a mutation in α-, β-, or γ-globin make the redox potential of the heme iron more negative; the oxidized heme then becomes more resistant to reduction by erythrocyte MetHb reductases [2, 13]. Consequently, Hb M disease causes methemoglobinemia and contributes to lifelong cyanosis that is unresponsive to oxygen therapy. In particular, the presence of cyanosis in the neonate supports an α-globin chain variant such as Hb M Boston, because of a β-globin mutation present several months later due to low β-chain expression at birth [2].
In this study, the range of SO2 on pulse oximetry was 77–83%. Although we applied supplemental oxygen, no significant increase in saturation was seen. By contrast, several estimated saturation values calculated from PaO2 using a blood gas analyzer were consistently above 95%. This discrepancy results from the unique effects of MetHb on standard oxygenation assessments and can distinguish it from cyanotic congenital heart disease [2, 4, 5]. MetHb can be measured directly in blood by co-oximetry using multiple wavelengths of light to distinguish not only the fractions of oxyhemoglobin and deoxyhemoglobin, but also MetHb and carboxyhemoglobin [3]. In some cases of Hb M, MetHb level can be underestimated when measured using co-oximetry [2, 5, 11, 14]. Certain mutations in the gene coding for one of the globin chains cause conformational changes in hemoglobin, and their absorbance spectrums can differ from typical MetHb [3]. Therefore, co-oximetry might not be useful to detect the percentage of MetHb arising from congenital variants like Hb M, as demonstrated in our case. In addition, unlike acquired methemoglobinemia with much higher amounts of MetHb, the patients with congenital methemoglobinemia typically have MetHb values of less than 20% that cause clinically obvious cyanosis; however, the patient is otherwise asymptomatic [4, 5].
In this study, the presence of Hb M disease and the pattern of de novo mutation were simultaneously established by DNA sequencing, not by electrophoresis or high-performance liquid chromatography. Because different Hb M variants may have similar profiles using some or all of these techniques, DNA gene sequencing is the definitive method for distinguishing various hemoglobin variants [2, 5].
Conclusion
Hb M disease is a rare blood disorder, but needs to be considered in newborns with cyanosis or low SO2 on pulse oximetry in the face of normal arterial blood saturation regardless of MetHb value. Early diagnosis helps to avoid unnecessary diagnostic approaches and aggressive interventions because the hemoglobin variants show a good prognosis.
Availability of data and materials
All available data is presented in the main manuscript.
Abbreviations
- Hb M:
-
Hemoglobin M
- MetHb:
-
Methemoglobin
- PaO2 :
-
Partial pressure of oxygen
- SO2 :
-
Oxygen saturation
References
Viana MB, Belisário AR. De novo alpha 2 hemoglobin gene (HBA2) mutation in a child with hemoglobin M Iwate and symptomatic methemoglobinemia since birth. Rev Bras Hematol Hemoter. 2014;36:230–4.
Estey MP, Clarke G, Sia W, Toor E, Higgins TN. A mother and newborn with brown blood. Clin Chem. 2015;61:466–9.
Haymond S. Oxygen saturation: a guide to laboratory assessment. Clin Lab News. 2006:10–2.
Nagel RL. Disorders of hemoglobin function and stability. In: Steinberg MH, forget BG, Higgs DR, et al. disorders of hemoglobin: genetics, pathophysiology and clinical management. Cambridge: Cambridge University Press; 2001. p. 1155–94.
Elboraee MS, Clarke G, Belletrutti MJ, Escoredo S. HbM methaemoglobinaemia as a rare case of early neonatal benign cyanosis. BMJ Case Rep. 2015; pii:bcr2015212336.
Gerald PS. The electrophoretic and spectroscopic characterization of Hgb M. Blood. 1958;13:936–49.
Gerald PS, Efron ML. Chemical studies of several varieties of Hb M. Proc Natl Acad Sci U S A. 1961;47:1758–67.
Jo I, Jang W, Chae H, Kim SY, Kim M, Kim Y, et al. Hemoglobin Kansas: first Korean family and literature review. Ann Lab Med. 2017;37:352–4.
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–75.
Mayne EE, Elder GE, Lappin TR, Ferguson LA. HB M Iwate [α287His→Tyrβ2]: de novo mutation in an Irish family. Hemoglobin. 1986;10:205–8.
Upadhye D, Koduri P, Tarakeshwari S, Mehta P, Surve R, Warang P, et al. Hb M Hyde Park and Hb M Boston in two Indian families – a rare cause of methaemoglobinemia. Int J Lab Hematol. 2015;37:e40–3.
Loong TY, Chong DL, Jamal AR, Murad NA, Sabudin RZ, Fun LC. First reported case of Haemoglobin-M Hyde Park in a Malay family living in Malaysia. EXCLI J. 2016;15:630–5.
Jin Y, Nagai M, Nagai Y, Nagatomo S, Kitagawa T. Heme structures of five variants of hemoglobin M probed by resonance Raman spectroscopy. Biochemistry. 2004;43:8517–27.
Alonso-Ojembarrena A, Lubián-López SP. Hemoglobin M disease as a cause of cyanosis in a newborn. J Pediatr Hematol Oncol. 2016;38:173–5.
Acknowledgements
The authors would like to thank the patient and their family for their patience and willingness to provide their case in the educational contribution.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
CWS and JHL drafted the manuscript and reviewed the literature. MSK performed the genetic studies of dyshemoglobin. CWS and MH gathered clinical information. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The case report was approved by the Institutional Review Board of St. Vincent’s Hospital. The IRB project number is VC18ZESI0133.
Consent for publication
Written informed consent was obtained from the patient’s parents for the publication of this case report. A copy of the notification of review and approval is available for review by the Editor of this journal.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shin, C., Hong, M., Kim, M. et al. Exon sequencing of the alpha-2-globin gene for the differential diagnosis of central cyanosis in newborns: a case report. BMC Pediatr 19, 221 (2019). https://doi.org/10.1186/s12887-019-1601-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1601-9