
Abstract
Background
Oculocutaneous albinism (OCA) is a genetically heterogeneous condition that is associated with reduced or absent melanin pigment in the skin, hair, and eyes, resulting in reduced vision, high sensitivity to light, and rapid and uncontrolled eye movements. To date, seventeen genes have been associated with OCA including syndromic and non-syndromic forms of the condition.
Methods
Whole exome sequencing (WES) was performed to identify pathogenic variants in nine Pakistani families with OCA, with validation and segregation of candidate variants performed using Sanger sequencing. Furthermore, the pathogenicity of the identified variants was assessed using various in-silico tools and 3D protein structural analysis software.
Results
WES identified biallelic variants in three genes explaining the OCA in these families, including four variants in TYR, three in OCA2, and two in HPS1, including two novel variants c.667C > T: p.(Gln223*) in TYR, and c.2009 T > C: p.(Leu670Pro) in HPS1.
Conclusions
Overall, this study adds further knowledge of the genetic basis of OCA in Pakistani communities and facilitates improved management and counselling services for families suffering from severe genetic diseases in Pakistan.
Similar content being viewed by others


Background
Albinism is a complex group of rare genetic disorders characterized by abnormal melanin biosynthesis, resulting in complete or partial loss of pigment (pheomelanin or eumelanin) with the addition of reduced visual acuity, photophobia (sensitivity to light), and nystagmus (random eye movements) [1]. It is divided into two major categories: ocular albinism (OA; MIM 300500), characterised by hypopigmentation of the ocular tissue, and oculocutaneous albinism (OCA; MIM 203100) (www.omim.org assessed on 30 January 2023) [2], which involves lack of pigmentation in the eyes, skin, and hair with nystagmus [3], misrouting of the optic nerves, foveal hypoplasia, and loss of vision. OCA is further classified into syndromic and non-syndromic forms. Non-syndromic OCA is caused by mutations in genes involved in melanin biosynthesis and melanocyte differentiation, resulting in only hypopigmentation and visual abnormalities [4]. Currently, seven genes (TYR, OCA2, TYRP1, SLC45A2, SLC24A5, LRMDA, and DCT) [5] linked to eight different types (OCA1-8) of non-syndromic OCA have been reported, with OCA1 being the most common, accounting for 50% of all cases reported worldwide associated with TYR gene variants [6]. Syndromic forms of OCA are associated with genes encoding proteins involved in the regulation of intercellular transport of molecules and the generation of lysosome-related organelles (LROs). LROs are specific to certain cell types, such as lytic granules in CD8 + T-cells and melanosomes in melanocytes. Disruption of this pathway can result in immunodeficiency, bleeding diathesis, and pulmonary fibrosis, as well as prominent OCA phenotypes such as hypopigmentation in the skin, eyes, and hair [7, 8]. Furthermore, syndromic OCA may include additional systemic changes. Hermansky-Pudlak syndrome (HPS; MIM 203300) and Chediak-Higashi syndrome (CHS; MIM 214500) are the two most common types of syndromic OCA. HPS is associated with mutations in genes involved in the formation of protein complexes (BLOC-1, BLOC-2, BLOC-3, or AP-3) and take part in biogenesis of specialized organelles such as melanosomes [9], whereas CHS is linked to mutations in the LYST gene located at 1q42-q43 and encodes a vascular transport protein whose function has not yet been fully delineated [10]. Clinical features of HPS generally include OCA with associated haematological problems (epistaxis, bleeding diathesis, menorrhagia, colonic and gingival bleeding and prolonged bleeding after surgery/trauma or postpartum hemorrhage), gastrointestinal anomalies (cramps, abdominal pain, enterocolitis, malabsorption, and diarrhoea), and respiratory issues (recurrent infection, exertional dyspnea, non-productive cough, pulmonary fibrosis, and hypoxia) [11]. To date, 11 HPS subtypes associated with 11 different genes have been described in the literature. Both syndromic and non-syndromic OCA are inherited in an autosomal recessive pattern and have a global prevalence that ranges from 1: 17,000 to 20,000, with nearly one in every 70 individuals being a carrier [12, 13]. However, prevalence varies based on OCA type, ethnicity, and distinct founder mutations present in specific populations.
This study details the molecular genetic analysis of nine consanguineous Pakistani families from different ethnic backgrounds with OCA and signs of nystagmus. Whole exome sequencing (WES) identified nine pathogenic variants in three protein-coding genes, including two novel variants in the TYR and HPS1 genes.
Methods
Ethical Approval
This study was approved by the International Islamic University's Institutional Review Board (IRB) in Islamabad, Pakistan (Letter No. IIU(BI&BT)/FBAS/2018/3598) and was carried out in accordance with the principles outlined in the Declaration of Helsinki. All participants in the study and/or their legal guardians provided written informed consent for clinical and research data to be published in a peer-reviewed journal. Nine affected families representing various ethnic groups were enrolled in this study from the Khyber Pakhtunkhwa (KPK) province of Pakistan. The clinical history of each family was recorded, and initial examination revealed the presence of OCA. Blood samples were collected from both affected and unaffected family members including their parents.
Clinical Examination
On initial analysis, complete family history and clinical information were obtained through questionnaire. The guardians of each of the families were interviewed about the onset of disease and all other related information. Pedigrees were constructed from information provided by the family using Cyrillic 2.0 software, according to the standard protocol defined by Bennett et al. (1995) [14]. After data collection, a detailed clinical evaluation was performed by a physician at the local district hospital for all affected and selected unaffected individuals from each of the nine families involved in the study to confirm their disease status. Colour vision analysis (Ishihara charts), visual acuity assessment (Snellen charts), photophobia, and fundoscopic examination by direct ophthalmoscopy were evaluated in affected individuals. Additionally, affected individuals were examined and assessed for pigment abnormalities in the skin, eyes, and hair to identify possible syndromic conditions associated with OCA.
Molecular genetic study
DNA was extracted from blood samples using an in-organic (salting out) protocol [15], carried out step by step at room temperature. Chemicals and reagents were kept at -4 °C and tightly sealed to avoid contamination. The NanoDrop™ spectrophotometer was used to determine the concentration and purity of the extracted DNA samples (Thermo Fisher Scientific, Dover, DE, USA).
WES was performed using an Illumina HiSeq™ 2000 sequencer on DNA from a single affected member from each of the nine families (Illumina Inc., San Diego, CA, USA). For exome enrichment, 51 Mb Agilent Sure Select Human All ExonV4 enrichment kit was used along with read alignment Burrows–Wheeler Aligner (BWA-MEM, v0.7.17) [16], InDel realignment, base quality recalibration Genome Analysis Tool Kit (GATK, v3.7.0) [17], SNVs/InDels (GATK/Haplotype Caller), duplicates removed and mate-pairs fixed using Picard (v2.15.0) (http://broadinstitute.github.io/picard/) and DNAnexus for annotation and variant calling (DNAnexus Inc., CA, USA: https://dnanexus.com/). Variant Call Format (VCF) files including all gene variations were generated using Haplotype Caller. Homozygosity was mapped using HomozygosityMapper [18]. The gnomAD database (https://gnomad.broadinstitute.org/) was used to detect allele frequencies, and GERP was used to conserve variants [19]. To identify candidate genes, single nucleotide polymorphisms (SNPs) with Minor Allele Frequency (MAF) (>0.05) were removed, along with non-splicing junctions containing synonymous and intronic variants found in the 1000 Genomes Project (www.1000genomes.org) [20] or the Single Nucleotide Polymorphism Database (dbSNP; NCBI).
Primers were designed using Primer3 software v0.4.0 (http://frodo.wi.mit.edu/primer3/) for all coding exons and associated intron–exon junctions of the TYR (NM_000372.5), OCA2 (NM_000275.3), and HPS1 (NM_000195.5) genes. To confirm co-segregation of the genetic variants identified by WES, PCR amplicons were generated in the Bio-Rad T100™ thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA) using gene-specific primers (Supplementary Table 1) employing standard optimization procedures [21] and purified using the BIGDYE® XTerminator™ Purification Kit (ABI, Applied Biosystems, Waltham, MA, USA), with dideoxy sequencing of amplicons using an ABI 3730 DNA Analyzer (Thermo Fisher Scientific, Dover, DE, USA). Sequence reads were aligned to the human genome reference sequence [hg38] to identify base changes using BioEdit 7.0 (http://www.mbio.ncsu.edu/BioEdit/bioedit.html), CLC sequence viewer 8.0 (http://www.clcbio.com/products/clc-sequence-viewer/) and chromatograms visualized with FinchTV v1.5.0 (https://digitalworldbiology.com/FinchTV) software. Reference sequences for TYR, OCA2, and HPS1 genes and proteins were obtained from the Ensemble genome browser (GRCh38 assembly, Dec 2013) (http://www.ensembl.org/Homo sapiens/Info/Index?db = core). Variant and allele frequencies were identified in ClinVar, HumVar, dbSNP, gnomAD v3.1.2, and HGMD 2022.1 online genomic databases, and the pathogenicity of TYR, OCA2, and HPS1 gene variants was determined using the ACMG/AMP [22] guidelines.
Bioinformatics analysis
In silico pathogenicity prediction tools were used to assess missense and splice variants including REVEL (rare exome variant ensemble learner) [23] scores taken from dbNSFP (v4.3 a) (http://database.liulab.science/dbNSFPconn) [24], PredictSNP2 (https://loschmidt.chemi.muni.cz/predictsnp2/) [25], Scale-Invariant Feature Transform (SIFT) (https://sift.bii.a-star.edu.sg/) [26], Polymorphism phenotyping v2 (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2/) [27], Protein Variation Effect Analyzer (PROVEAN) (https://www.jcvi.org/research/provean) [28], SpliceAI (https://spliceailookup.broadinstitute.org/) [29] and NNsplice (https://www.fruitfly.org/seq_tools/splice.html) [30].
Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [31] was employed to show protein conservation across several species and for a three-dimensional (3D) structural analysis, the normal and mutant protein structures were generated using different protein prediction software’s such as AlphaFold (https://alphafold.ebi.ac.uk/) [32], SWISS-MODEL (https://swissmodel.expasy.org/) [33], Phyre2 v2.0 (http://www.sbg.bio.ic.ac.uk/~phyre2/) [34], and RoseTTAFold (https://robetta.bakerlab.org/) [35]. Ramachandran plots were used to evaluate the stereochemistry and validity of the constructed 3D protein structures [36]. ERRAT [37], VERIFY 3D [38], WHATCHECK [39], and PROCHECK [36] evaluation tools (https://saves.mbi.ucla.edu/) were used for the assessment and verification of the predicted structures and then suitable structures were chosen based on their preferred regions and the ERRAT quality factor. Structures were analyzed using the Swiss-Pdb viewer (https://spdbv.unil.ch/) [40] to check for variant effect on protein structure. Furthermore, structure refinement, energy minimization, and visualization were done by UCSF Chimera 1.16 (http://www.cgl.ucsf.edu/chimera) [41].
Results
Clinical description
Nine consanguineous Pakistani families with OCA were recruited from Pakistan's KPK province. In all families, pedigree analysis revealed a recessive pattern of inheritance (Fig. 1). All affected individuals from the nine families showed signs of nystagmus and decreased visual acuity with depigmentation of the hair, eyes, and skin, whereas, the presence of photophobia, strabismus, colour blindness, and foveal hypoplasia differed between individuals. In families PKNYS 08 and 09, additional symptoms of epistaxis, gingival bleeding, and bruising with severe and frequent respiratory infections were observed in affected individuals IV:1 from each family respectively, indicating the presence of a syndromic form of OCA (HPS). Detailed clinical evaluation is described in Table 1.

Pedigrees of families. PKNYS (01–04) with OCA co-segregating for TYR mutations, PKNYS (05–07) with OCA co-segregating for OCA2 mutations, and families PKNYS (08,09) with OCA co-segregating for HPS1 mutations. “ + ” sign indicates wildtype allele whereas “-” sign indicates mutated allele. For compound heterozygous mutations, the different TYR variants within the same family are displayed in different colours
Genetic findings
Variants in TYR
WES identified four pathogenic TYR variants in families PKNYS (01–04), including a novel nonsense variant [Chr11(GRCh38):g.89178620C > T; NM_000372.5: c.667C > T; p.(Gln223*)] in the first exon of TYR in family PKNYS01, resulting in a premature termination codon (PTC) and truncated protein predicted to undergo nonsense-mediated decay (NMD). The MAF for the variant is not indexed in gnomAD V3.1.2 and is listed as ‘pathogenic’ in ClinVar, however, there is no mention of zygosity. Furthermore, three previously reported [42,43,44] TYR variants were identified including a missense mutation in exon one [Chr11(GRCh38):g.89178085 T > A; NM_000372.5: c.132 T > A; p.(Ser44Arg)] in family PKNYS02, a nonsense mutation [Chr11(GRCh38):g.89191214C > T; NM_000372.5: c.832C > T; p.(Arg278*)] in family PKNYS03, and compound heterozygous variants in family PKNYS04 [NM_000372.5: Chr11(GRCh38):g.89191215C > T; c.832C > T; p.(Arg278*); and Chr11(GRCh38):g.89284843G > A; c.1255G > A; p.(Gly419Arg)] in exons one and four respectively. All variants co-segregated in families PKNYS 01–04 as expected for an autosomal recessive condition (see Fig. 1). All the identified variants were predicted to alter the function/expression of the TYR gene (Table 2).
Variants in OCA2
WES identified two previously reported variants [12, 45] in the OCA2 gene in families PKNYS (05–07). The frameshift variant [Chr15(GRCh38):g.28027977_28027978DelTT; NM_000275.3: c.408_409delAA; p.(Arg137Ilefs*83)] located in exon four in family PKNYS05 and a splice site variant [Chr15(GRCh38):g.27990662A > C; NM_000275.3: c.1045-15 T > G; p.?] in families PKNYS 06 and 07 resulting in skipping of exon ten. All variants co-segregated as expected for an autosomal recessive condition in families PKNYS (05–07) (Fig. 1). The identified mutations are predicted to alter OCA2 expression in the affected individuals resulting in the disease phenotype.
Variants in HPS1
WES identified one novel and one previously reported mutation in the HPS1 gene in families PKNYS 08 and 09. The novel missense variant [Chr10(GRCh38):g.98417658A > G; NM_000195.5: c.2009 T > C; p.(Leu670Pro)] was located in exon twenty of HPS1 in family PKNYS09. The MAF for the variant is not indexed in gnomAD V3.1.2 and is listed as ‘likely pathogenic’ in ClinVar, however, no clinical details are provided. The previously reported [46] nonsense mutation [Chr10(GRCh38):g.98431282G > A; NM_000195.5: c.517C > T; p.(Arg173*)] in exon seven in family PKNYS08. Co-segregation analysis in families PKNYS 08 and 09 revealed parents as heterozygous carriers and affected individuals as homozygous for these HPS1 variants as expected for an autosomal recessive condition (Fig. 1).
In-Silico analysis
Various online tools were used to predict the pathogenicity of identified missense variants, including REVEL score, PredictSNP2, SIFT, PolyPhen-2, and PROVEAN. SpliceAI and NNSPLICE were used to assess the effect of splice variants. Table 2 details the results of these pathogenicity prediction tools alongside HGMD variant classification. SIFT, PolyPhen-2, PROVEAN, PredictSNP2, and Mutation Taster scores for the novel HPS1 variant (NM_000195.5: c.2009 T > C; p.(Leu670Pro)) were 0.000, 0.998, -5.789, 1.000, and 0.999 respectively, and all predict the mutation to be deleterious.
Structural analysis
To investigate the effect and relationship between the wild-type (WT) and mutant protein structures for the novel TYR and HPS1 variants, 3D-structural analysis was performed. It was predicted that the mutant TYR protein resulting from the nonsense variant (NM_000372.5: c.667C > T; p.Gln223*) would/may produce a truncated structure of only 222 amino acids in length (Fig. 2, Supplementary Fig. 1), caused by a PTC as compared to the WT TYR protein comprising of 529 residues (Supplementary Fig. 1). Furthermore, Ramachandran plots of the WT TYR protein revealed 88.3% and 10.9% residues in the favored and allowed regions, while the mutant TYR protein consisting of only 222 amino acids (less than half of WT) had 88.9% and 11.9% residues in the favored and allowed regions (Supplementary Fig. 1). WT and mutant TYR structures were further analyzed in Swiss-Pdb viewer. The p. Gln223* termination was in the intra-melanosome domain of the TYR protein and resulted in a truncated non-functional protein structure For the HPS1 protein (700 residues), the missense variant (NM_000195.5: c.2009 T > C) was predicted to cause a substitution of amino acid leucine to proline at position 670 (Supplementary Fig. 2). The WT and mutant HPS1 proteins were superimposed for direct comparison revealing conformational changes and altered quaternary structure of the mutant HPS1 protein (Fig. 3C, D). Additionally, Ramachandran plots for the WT HPS1 protein showed 87.8% and 11.2% residues in the favored and allowed regions, whereas the mutant HPS1 protein had 87.6% and 11.9% residues in the favored and allowed regions (Supplementary Fig. 2. WT and mutant HPS1 structures were then analyzed in Swiss-Pdb viewer. The p.Leu670Pro substitution was revealed to be located in C-terminal Fuz-longin-3 domain of the HPS1 protein, which takes part in Rab signalling (Figs. 3 and 4) by forming a complex (BLOC-3) with HPS3. The substitution resulted in alteration of quaternary protein structure and predicted to disturb Rab signalling via BLOC-3. Overall, the reported pathogenic variants resulted in defective/altered protein structures leading to the OCA phenotype in affected individuals.

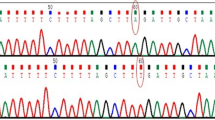
Genetic analysis of family PKNYS01 with novel TYR variant. A From top to bottom: chromatograms of unaffected wildtype individual (Top), unaffected heterozygous carrier (Centre), and affected individual homozygous for thymine at position c.667. B Normal TYR structure consisting of five exons (1–5) which encode for the essential signal sequence, intra-melanosomal domain (consisting of epidermal growth factor (EGF)-like region and a Copper (Cu)-containing domains), and transmembrane-domain (Top); Mutated TYR structure showing stop codon at position p. Gln 223* in the intra-melanosomal domain along with scale for amino acid length (Bottom)

Genetic analysis of family PKNYS09 with novel HPS1 variant. A From top to bottom: chromatograms of unaffected wildtype individual (Top), unaffected heterozygous carrier (Centre), and affected individual homozygous for cytosine at position c.2009. B ClustalO multiple amino acid sequence alignment of HPS1 orthologs shows p.Leu670 as highly conserved among species (shaded area represent conserved amino acids; light area represent non-conserved amino acids)

Protein modelling studies (Swiss model, SPDBV v4.10) demonstrating the location of the HPS1 670Leu position within the C-terminal Fuz-longin-3 domain (Third Longin domain of FUZ, MON1 and HPS1, orange) [47, 48]. Structurally this domain is composed of an α/β fold which contains five anti-parallel β-strands organised as a central β-sheet, with two α-helices around it (Sanchez-Pulido & Ponting, 2020). (A, B) demonstrate the reference amino acid Leucine and (C, D) show the alternate Proline arising from the novel HPS1 variant (NM_000195.5: c.2009 T > C; p.(Leu670Pro)). The location of this variant within the central β-sheet is likely to affect the function of this domain in Rab signalling
Discussion
OCA is a clinically and genetically heterogeneous disorder that has been observed to segregate in an autosomal recessive pattern in humans [13]. This study identifies pathogenic variants in TYR, OCA2, and HPS1 genes in affected individuals from nine Pakistani families with OCA (Table 1) portraying a recessive form of inheritance in all cases (Fig. 1). A total of nine mutations were identified using WES, including two novel variants in the TYR and HPS1 genes (Table 2), moreover, in-silico analysis of altered amino acid sequences and 3D structural prediction further support the pathogenicity of these variants (Figs. 2, 3, 4 and Supplementary Fig. 1,2).
The TYR (NM_000372.5) gene located at chromosome 11q14.3 consists of five exons encoding an enzyme 523 amino acid residues in length which catalyzes the first two steps of the melanin biosynthesis pathway and involves oxidation and hydroxylation of L-DOPA and DOPA quinone. The glycoprotein structure consists of four regions, a signal sequence (1‑18), an intra-melanosomal domain (19‑476) comprising of a copper binding site, a single α‑helical trans‑membrane domain (477‑497), and a flexible C‑terminal domain (498‑529) [49]. To date, over 565 pathogenic variants have been reported in the TYR gene (HGMD 2022.1) (Table 3) with over 90 mutations identified in the Pakistani population, accounting for 40% of OCA cases. Furthermore, the prevalence of TYR allele in the Pakistani population accounts for about 37% [50]. In families PKNYS (01–04) four mutations (three previously reported, one novel) were identified in the TYR gene. Among the reported variants, c.132 T > A (p.Ser44Arg) has been previously described in three Pakistani families and is more common in the South Asian population according to gnomAD v2.1.2 MAF (0.0013), compared to other populations (African 0.0001249, and European 0.00002372) [51]. The TYR variants (p.Gly419Arg) c.1255G > A and (p.Arg278*) c.832C > T are amongst the most frequently reported mutations in the Pakistani community occurring in 20 and 21 families, respectively [5]. The reported nonsense/missense variants c.132 T > A, c.832C > T and c.1255G > A (p.Ser44Arg, p.Arg278* and p.Gly419Arg) were found to be rare with MAF 0.0001, 0.0001, and 0.00006 worldwide respectively [50] whereas, MAF for the novel TYR nonsense variant c.667C > T (p.Gln223*) was not listed in gnomAD v3.1.2. The c.667C > T; p.(Gln223*) variant is listed as pathogenic in ClinVar, although there are limited clinical details and no information on zygosity. The variant results in a truncated non-functional structure occurring in the first exon encoding the intra-melanosomal domain (IMD) (conserved region),which facilitate the enzymatic conversion of the amino acid tyrosine into melanin. The IMD of tyrosinase contains the active site of the enzyme, where it catalyzes two key reactions in the melanin synthesis pathway: (1) Hydroxylation of Tyrosine and (2) Oxidation of DOPA. These enzymatic reactions are crucial in melanin biosynthesis. Alterations in the IMD results in loss of TYR function leading to genetic conditions such as OCA [52] like in family PKNYS01. In families PKNYS (05–07) two previously reported mutations were identified in the OCA2 gene responsible for OCA type 2 (second most prevalent form of OCA), which has a global prevalence of 1 in 36,000. The OCA2 (NM_000275.3) gene previously known as the ‘P’ gene is composed of 23 exons and codes for a melanosomal transmembrane enzyme consisting of 838 residues (10 kDa) [53]. Until now, more than 431 mutations have been reported in the OCA2 gene (HGMD 2022.1) (Table 3), with a total of 59 variants reported in the Pakistani population in seven studies. MAF for the identified OCA2 mutations c.408_409delAA (p.Arg137Ilefs*83) and c.1045-15 T > G (splice site mutation) in families PKNYS (05, and 06/07) shows not listed and 0.00002 respectively in gnomAD v3.1.2 [51]. The splice site variant c.1045-15 T > G although very rare has been widely reported in more than 17 families belonging to the Pakistani population accounting for over 30% of the total mutations in the OCA2 gene [50] whereas, the frameshift variant c.408_409delAA (p.Arg137Ilefs*83) identified in family PKNYS05 has only been reported once before in a single Pakistani family [12].
In addition to the OCA phenotype, the affected individuals (IV:1) from families PKNYS 08 and 09 showed symptoms like epistaxis and bruising accompanied by infections, indicating the presence of HPS (syndromic OCA). WES identified one novel and one previously reported variant in the HPS1 gene in affected individuals. The HPS1 (NM_000195.5) gene responsible for HPS type 1 is located on the reverse strand of chromosome 10q23.1-q23.3 and codes for a 700-residue protein structure [54], which plays a crucial role in melanosome regulation, organelle biogenesis, and has been reported to interact with TYR, TYRP1 and TRP2/DCT. HPS1 affects individuals of different ethnicities, including those from European, Asian, and South American backgrounds. It has a global prevalence of 1/1,500,000–1,000,000, although the prevalence is 1/1800 in individuals of Puerto Rican decent [55]. Over 98 mutations have been reported in the HPS1 gene (HGMD 2022.1), with 9 variants identified in the Pakistani population in four reports [11] from all HPS subtypes, of which three mutations were present in the HPS1 gene (c.1342 T > C, genomic deletion, c.2056C > T) [56]. The reported variant c.517C > T (p.Arg173*) in family PKNYS08 has been previously described in the Chinese population [46] and although is rare with a MAF of 0.00003, whereas the MAF for the novel mutation c.2009 T > C (p.Leu670Pro) is not listed in gnomAD v3.1.2, and this variant is listed as likely pathogenic in Clinvar, although no evidence or clinical details are provided. The variant lies in the C-terminal Fuz-longin-3 domain (conserved region; Figs. 3 and 4) of the HPS1 protein which forms a complex (BLOC-3) with HPS4. This complex acts as a guanine nucleotide exchange factor (GEF) and shows specific activity toward Ra32/38 and can promote recruitment of Rab32 and Rab38 to membrane. Furthermore, BLOC-3 and its target Rabs act in the biogenesis of melanosomes and alterations in these complexes have been reported to cause syndromic forms of OCA like HPS [47, 48]. The resulting substitution p.Leu670Pro induces changes in the fuzz-domain of HPS1 protein altering the quaternary structure due to different characteristics of the amino acids leading to changes in Rab signalling (Fig. 4) thus, supporting the pathogenicity of this variant in family PKNYS09. Currently, there is no potential treatment for OCA, and management strategies focus on proper eye care and monitoring skin for problems.
Conclusion
Our study identifies novel and reported variants in the TYR, OCA2, and HPS1 genes and broadens the mutational spectrum and genetic heterogeneity of OCA in the Pakistani population. We further predict the deleterious outcome of novel variants using in-silico variant prediction tools and structural analysis. These findings will assist in providing an early diagnosis for affected individuals and facilitate the provision of possible genetic counselling for affected families in the Pakistani community and worldwide.
Availability of data and materials
The patient’s non-sensitive datasets used and/or analysed during the current study are available from the corresponding authors on reasonable request.
References
Shah S, Saeed A, Irshad M, Babar M, Hussain T. Oculocutaneous albinism in Pakistan: A review. J Cancer Sci Ther. 2018;10:253–7.
Rocca C, Tiberi L, Bargiacchi S, Palazzo V, Landini S, Marziali E, Caputo R, Tinelli F, Marchi V, Benedetto A. Expanding the Spectrum of Oculocutaneous Albinism: Does Isolated Foveal Hypoplasia Really Exist? Int J Mol Sci. 2022;23:7825.
Arshad MW, Shabbir MI, Asif S, Shahzad M, Leydier L, Rai SK. FRMD7 Gene Alterations in a Pakistani Family Associated with Congenital Idiopathic Nystagmus. Genes. 2023;14(2):346.
Schidlowski L, Liebert F, Iankilevich PG, Rebellato PRO, Rocha RA, Almeida NAP, Jain A, Wu Y, Itan Y, Rosati R. Non-syndromic oculocutaneous albinism: novel genetic variants and clinical follow up of a brazilian pediatric cohort. Front Genet. 2020;11:397.
Ullah MI. Clinical and Mutation Spectrum of Autosomal Recessive Non-Syndromic Oculocutaneous Albinism (nsOCA) in Pakistan: A Review. Genes. 2022;13:1072.
Shakil M, Akbar A, Aisha NM, Hussain I, Ullah MI, Atif M, Kaul H, Amar A, Latif MZ, Qureshi MA. Delineating Novel and Known Pathogenic Variants in TYR, OCA2 and HPS-1 Genes in Eight Oculocutaneous Albinism (OCA) Pakistani Families. Genes. 2022;13:503.
Fernández A, Hayashi M, Garrido G, Montero A, Guardia A, Suzuki T, Montoliu L. Genetics of non-syndromic and syndromic oculocutaneous albinism in human and mouse. Pigment Cell Melanoma Res. 2021;34:786–99.
Bibi N, Ullah A, Darwesh L, Khan W, Khan T, Ullah K, Khan B, Ahmad W, Umm-e-Kalsoom. Identification and computational analysis of novel tyr and slc45a2 gene mutations in Pakistani families with identical non-syndromic oculocutaneous Albinism. Front Genet. 2020;11:749.
El-Chemaly S, Young LR. Hermansky-pudlak syndrome. Clin Chest Med. 2016;37:505–11.
Ajitkumar A, Yarrarapu SNS, Ramphul K. Chediak Higashi Syndrome. 2018.
Zaman Q, Anas M, Rehman G, Khan Q, Iftikhar A, Ahmad M, Owais M, Ahmad I, Muthaffar OY, Abdulkareem AA. Report of Hermansky-Pudlak Syndrome in Two Families with Novel Variants in HPS3 and HPS4 Genes. Genes. 2023;14:145.
Arshad MW, Harlalka GV, Lin S, D’Atri I, Mehmood S, Shakil M, Hassan MJ, Chioza BA, Self JE, Ennis S. Mutations in TYR and OCA2 associated with oculocutaneous albinism in Pakistani families. Meta Gene. 2018;17:48–55.
Sajid Z, Yousaf S, Waryah YM, Mughal TA, Kausar T, Shahzad M, Rao AR, Abbasi AA, Shaikh RS, Waryah AM. Genetic causes of oculocutaneous albinism in Pakistani population. Genes. 2021;12:492.
Bennett RL, Steinhaus KA, Uhrich SB, O’Sullivan CK, Resta RG, Lochner-Doyle D, Markel DS, Vincent V, Hamanishi J. Recommendations for standardized human pedigree nomenclature. J Genet Couns. 1995;4:267–79.
Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17:8390.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic acids research. 2009;37(suppl_2):W593–9.
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS computational biology. 2010;6(12):e1001025.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, MacArthur DG, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-43.
Asif S, Khan M, Arshad MW, Shabbir MI. PCR Optimization for Beginners: A Step by Step Guide. Research in Molecular Medicine. 2021;9:81–102. https://doi.org/10.32598/rmm.9.2.1189.1.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. The American Journal of Human Genetics. 2016;99:877–85.
Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome medicine. 2020;12:1–8.
Bendl J, Musil M, Štourač J, Zendulka J, Damborský J, Brezovský J. PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput Biol. 2016;12: e1004962.
Lowe DG. Object recognition from local scale-invariant features. In: Proceedings of the Proceedings of the seventh IEEE international conference on computer vision. 1999. p. 1150–7.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Jaganathan K, Panagiotopoulou SK, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(535–548): e524.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. In: Proceedings of the Proceedings of the first annual international conference on Computational molecular biology. 1997. p. 232–40.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–303.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–58.
Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–6.
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–91.
Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–9.
Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–5.
Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272–272.
Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modelin.g. Electrophoresis. 1997;18(15):2714–23.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
Shah S, Raheem N, Daud S, Mubeen J, Shaikh A, Baloch A, Nadeem A, Tayyab M, Babar M, Ahmad J. Mutational spectrum of the TYR and SLC45A2 genes in Pakistani families with oculocutaneous albinism, and potential founder effect of missense substitution (p. Arg77Gln) of tyrosinase. Clin Exp Dermatol. 2015;40:774–80.
Wang X, Zhu Y, Shen N, Peng J, Wang C, Liu H, Lu Y. Mutation analysis of a Chinese family with oculocutaneous albinism. Oncotarget. 2016;7:84981.
Chaki M, Sengupta M, Mondal M, Bhattacharya A, Mallick S, Ray K. Molecular and functional studies of tyrosinase variants among Indian oculocutaneous albinism type 1 patients. Differentiation. 2011;22:2992–3003.
Jaworek TJ, Kausar T, Bell SM, Tariq N, Maqsood MI, Sohail A, Ali M, Iqbal F, Rasool S, Riazuddin S. Molecular genetic studies and delineation of the oculocutaneous albinism phenotype in the Pakistani population. Orphanet J Rare Dis. 2012;7:1–19.
Wei A, Yuan Y, Bai D, Ma J, Hao Z, Zhang Y, Yu J, Zhou Z, Yang L, Yang X. NGS-based 100-gene panel of hypopigmentation identifies mutations in Chinese Hermansky-Pudlak syndrome patients. Pigment Cell Melanoma Res. 2016;29:702–6.
Gerondopoulos A, Langemeyer L, Liang J-R, Linford A, Barr FA. BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr Biol. 2012;22(22):2135–9.
Sanchez-Pulido L, Ponting CP. Hexa-Longin domain scaffolds for inter-Rab signalling. Bioinformatics. 2020;36(4):990–3.
Sun W, Shen Y, Shan S, Han L, Li Y, Zhou Z, Zhong Z, Chen J. Identification of TYR mutations in patients with oculocutaneous albinism. Mol Med Rep. 2018;17:8409–13.
Shakil M, Harlalka GV, Ali S, Lin S, D’Atri I, Hussain S, Nasir A, Shahzad MA, Ullah MI, Self JE. Tyrosinase (TYR) gene sequencing and literature review reveals recurrent mutations and multiple population founder gene mutations as causative of oculocutaneous albinism (OCA) in Pakistani families. Eye. 2019;33:1339–46.
Shahzad M, Yousaf S, Waryah YM, Gul H, Kausar T, Tariq N, Mahmood U, Ali M, Khan MA, Waryah AM. Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population. Sci Rep. 2017;7:44185.
Farney SK, Dolinska MB, Sergeev YV. Dynamic analysis of human tyrosinase intra-melanosomal domain and mutant variants to further understand oculocutaneous albinism type 1. Journal of analytical & pharmaceutical research. 2018;7(6):621.
Donnelly MP, Paschou P, Grigorenko E, Gurwitz D, Barta C, Lu R-B, Zhukova OV, Kim J-J, Siniscalco M, New M. A global view of the OCA2-HERC2 region and pigmentation. Hum Genet. 2012;131:683–96.
Huizing M, Malicdan MCV, Gochuico BR, Gahl WA. Hermansky-pudlak syndrome. 2021.
Rojas WDJ, Young LR. Hermansky–Pudlak syndrome. In: Proceedings of the Seminars in Respiratory and Critical Care Medicine. 2020. p. 238–46.
Yousaf S, Shahzad M, Tasleem K, Sheikh SA, Tariq N, Shabbir AS, Ali M, Waryah AM, Shaikh RS, Riazuddin S. Identification and clinical characterization of Hermansky-Pudlak syndrome alleles in the Pakistani population. Pigment Cell Melanoma Res. 2016;29:231.
Acknowledgements
We are thankful to all the individuals and their families for participating in this study. We also thank all clinicians and geneticists that helped us to perform this study in any shape specially Lyton-Rehmatullah Benevolent Trust (LRBT), and Christian Hospital, Quetta, Pakistan.
Funding
This study was partially supported by the Higher Education Commission (HEC) of Pakistan by awarding International Research Support Initiative Program (IRSIP) (Grant No: 1–8/HEC/HRD/2017/8347, PIN: IRSIP 39 BMS 1) to J.K and RILD Wellcome Wolfson Centre (Level 4), Royal Devon and Exeter NHS Foundation Trust, UK. Exome/Sanger sequencing and analysis was carried out at RILD Wellcome Wolfson Center UK and was funded by HEC Pakistan and Wellcome Trust UK (to E.L.B).
Author information
Authors and Affiliations
Contributions
Conceptualization, J.K and M.I.S; methodology, J.K, S.G, H.K, S.A.K and M.W.A; software, S.A and M.I.S; validation, S.A; formal analysis, J.K; investigation, J.K, C.G.S and S.L; resources, M.I.S, E.L.B, and A.H.C; data curation, J.K, S.A, L.E.R and M.I.S; writing—original draft preparation, J.K, S.A and M.I.S; writing—review and editing All Authors; supervision, E.L.B, A.H.C and M.I.S; project administration, M.I.S.; funding acquisition, E.L.B and A.H.C; All authors have read and approved the final version of the manuscript.
Authors information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Institutional Review Board (IRB) of the International Islamic University, Islamabad, Pakistan (Letter No. IIU(BI&BT)/FBAS/2018/3598), and the study was carried out in accordance with the principles outlined in the Declaration of Helsinki. Written informed consent were obtained from individuals > 18 years and from parents/guardians of individuals < 18 year participated in this study.
Consent for publication
Not Applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12886_2024_3611_MOESM1_ESM.docx
Supplementary Material 1. Supplementary Figure 1 3D structure of TYR protein. (A) wild type TYR from UniProt ID: P14679. (B) mutant TYR truncated at p. Q223* predicted using RoseTTAFold (C) Ramachandran plot of wild type TYR protein model (D) Ramachandran plot of Q223* mutant TYR protein structure.
12886_2024_3611_MOESM2_ESM.docx
Supplementary Material 2. Supplementary Figure 2 Superimposed 3D structure. (A) Wild type HPS1 (light blue) from UniProt ID: Q92902 and mutant HPS1 (purple) predicted using RoseTTAFold (B) Analysis of wildtype and mutant HPS1 proteins: (left) normal HPS1 structure depicting Leu670 in yellow; (right) altered HPS1 protein showing Pro670 substitution in red (C) Ramachandran plot of wild type HPS1 protein structure (D) Ramachandran plot of L670P mutant HPS1 protein model.
12886_2024_3611_MOESM3_ESM.docx
Supplementary Material 3. Supplementary Table 1 List of specific primer sequences for TYR, OCA2, and HPS1 genes used for co-segregation analysis in families with OCA.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, J., Asif, S., Ghani, S. et al. Mutational spectrum associated with oculocutaneous albinism and Hermansky-Pudlak syndrome in nine Pakistani families. BMC Ophthalmol 24, 345 (2024). https://doi.org/10.1186/s12886-024-03611-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-024-03611-6