
Abstract
Background
Familial exudative vitreoretinopathy (FEVR) is a genetic eye disorder that leads to abnormal development of retinal blood vessels, resulting in vision impairment. This study aims to identify pathogenic variants by targeted exome sequencing in 9 independent pedigrees with FEVR and characterize the novel pathogenic variants by molecular dynamics simulation.
Methods
Clinical data were collected from 9 families with FEVR. The causative genes were screened by targeted next-generation sequencing (TGS) and verified by Sanger sequencing. In silico analyses (SIFT, Polyphen2, Revel, MutationTaster, and GERP + +) were carried out to evaluate the pathogenicity of the variants. Molecular dynamics was simulated to predict protein conformation and flexibility transformation alterations on pathogenesis. Furthermore, molecular docking techniques were employed to explore the interactions and binding properties between LRP5 and DKK1 proteins relevant to the disease.
Results
A 44% overall detection rate was achieved with four variants including c.4289delC: p.Pro1431Argfs*8, c.2073G > T: p.Trp691Cys, c.1801G > A: p.Gly601Arg in LRP5 and c.633 T > A: p.Tyr211* in TSPAN12 in 4 unrelated probands. Based on in silico analysis and ACMG standard, two of them, c.4289delC: p.Pro1431Argfs*8 and c.2073G > T: p.Trp691Cys of LRP5 were identified as novel pathogenic variants. Based on computational predictions using molecular dynamics simulations and molecular docking, there are indications that these two variants might lead to alterations in the secondary structure and spatial conformation of the protein, potentially impacting its rigidity and flexibility. Furthermore, these pathogenic variants are speculated to potentially influence hydrogen bonding interactions and could result in an increased binding affinity with the DKK1 protein.
Conclusions
Two novel genetic variants of the LRP5 gene were identified, expanding the range of mutations associated with FEVR. Through molecular dynamics simulations and molecular docking, the potential impact of these variants on protein structure and their interactions with the DKK1 protein has been explored. These findings provide further support for the involvement of these variants in the pathogenesis of the disease.
Similar content being viewed by others


Introduction
Familial exudative vitreoretinopathy (FEVR) is a highly heterogeneous and monogenic genetic disorder that affects the development of retinal blood vessels. FEVR presents with diverse signs such as peripheral retina perfusion deficiency, neovascularization, vitreoretinal traction, macular displacement, retinal folds, and retinal detachment (RD). Interestingly, family members carrying the same pathogenic variant may display varying clinical features, including complete blindness, or remain asymptomatic. Nonetheless, this disease has not exhibited gender differences [1,2,3]. However, due to the incomplete penetrance of the disease and the similarities in fundus manifestations with other retinal diseases, such as high myopia or retinopathy of prematurity (ROP), the disorder can be prone to underdiagnosis or misdiagnosis [4,5,6]. Therefore, a combination of fundus imaging and genetic screening is crucial for the diagnosis and genetic counseling of FEVR.
FEVR exhibits various inheritance patterns, including autosomal dominant (AD), autosomal recessive (AR), and X-linked inheritance [7]. Several genes have been identified in individuals affected by familial exudative vitreoretinopathy (FEVR). These genes encompass low-density lipoprotein receptor-related protein 5 (LRP5), frizzled-4 (FZD4), tetraspanin-12 (TSPAN12), zinc finger protein 408 (ZNF408), Norrie disease protein (NDP), catenin beta 1 (CTNNB1), exudative vitreoretinopathy 3 (EVR3), kinesin family member 11 (KIF11), atonal homolog 7 (ATOH7), RCC1 and BTB domain-containing protein 1 (RCBTB1), and jagged canonical Notch ligand 1 (JAG1). Significantly, the FZD4, LRP5, TSPAN12, and NDP genes play crucial roles in retinal vascular development through the Norrin-β-catenin (Wnt) signaling pathway, collectively accounting for approximately 90% of molecularly diagnosed FEVR cases. It's noteworthy that only 50% of FEVR cases feature identifiable variants, with the remaining 50% potentially linked to yet undiscovered loci or genes [8,9,10,11,12]. Therefore, the detection of these genetic variants is crucial for precise diagnosis and understanding of the underlying pathogenesis of the disease.
Although several studies have confirmed the pathogenicity of variants in FEVR patients using second-generation sequencing technologies and in silico analysis, few attempts have been made to analyze the conformational changes of protein structures at the atomic or molecular level. Molecular dynamics (MD) simulations have been utilized in other ophthalmologic studies to investigate the effects of mutant proteins on conformational dynamics [13,14,15,16,17], underscoring the significance of this approach in comprehending the functional mechanisms of proteins and the molecular underpinnings of diseases [18].
To explore the pathogenicity of FEVR variants at the dynamic molecular level, we combined targeted next-generation sequencing (TGS) panel with MD simulations and molecular docking to characterize novel pathogenic variants in nine Chinese families with clinically diagnosed FEVR. This approach aimed to provide additional evidence for understanding the molecular basis of FEVR.
Methods
Study subjects and clinical examinations
Clinical data were collected from patients with a confirmed diagnosis of FEVR who were treated at the Ningxia Eye Hospital and Ningxia Hui Autonomous Region People's Hospital. The study was conducted by the Declaration of Helsinki and approved by the Ethics Committee of Ningxia Hui Autonomous Region People's Hospital (No. 2020-KY-GZR019). Each participant or their legal guardians provided informed consent before participating in the study.
All probands and family members underwent comprehensive ophthalmic examinations, which included assessment of best corrected visual acuity (BCVA), slit-lamp biomicroscopy, funduscopy, ultra-widefield fundus photography (UWFFP, Daytona P200T, OPTOS, Fife, UK), and fundus fluorescein angiography (FFA; Spectralis HRA + OCT, Heidelberg Engineering GmbH, Heidelberg, DE). FEVR was diagnosed in patients presenting with avascular peripheral retina and neovascularization, brush-like vessels, RD, vitreous hemorrhage, severe subretinal exudates, and macular ectopia, as per the criteria outlined in previous studies [19]. The disease stage in patients with FEVR was determined for each eye using Trese's staging system [3]. Additionally, basic patient information, such as gender, age, birth history, and family history, was collected. Patients with a history of premature delivery or other retinal vascular diseases with similar clinical manifestations were excluded from the study.
Pathogenic variant identification and in silico analysis
In this study, blood samples were collected from the proband and their family members to conduct TGS. Genomic DNA was extracted from peripheral blood leukocytes using the BaiMeng Magnetic Bead-based DNA Extraction Kit (BaiMeng, China). A whole-genome library was prepared using the NadPrep Whole-Genome Library Construction Kit (Swift Biosciences, USA) and selective enrichment of the exon regions of 511 target genes was performed using the SureSelect XT HS2 kit (Agilent Technologies, USA). Subsequently, paired-end 150-bp sequencing was performed on an Illumina HiSeq 2500 platform (Illumina, USA). The clean reads were aligned to the human reference genome hg19 (GRCh37) using the Burrows-Wheeler Aligner (BWA) software (Broad Institute of MIT and Harvard, USA). PCR duplicates were removed, and base quality score recalibration and variant calling were performed using the Genome Analysis Toolkit (GATK) software suite (Broad Institute, USA). The detected variants were annotated using the ANNOVAR software package (University of Michigan, USA) with data from various public databases, including dbSNP [20], ExAC [21], gnomAD [22], and 1000genomes [23]. Next, we excluded synonymous variants and non-coding region variants that had no impact on splicing signals. Additionally, variants with a high minor allele frequency of 0.01 or greater were also omitted from consideration. The pathogenicity and protein function of the variants were predicted using various tools such as SIFT [24], MutationTaster [25], PolyPhen-2 [26], REVEL [27], and GERP + + [28, 29]. Each variant was further filtered in ClinVar [30] and HGMD [31] databases to confirm whether it was previously reported. Subsequently, mutations within LRP5 (referenced by NM_002335.4 for mRNA) and TSPAN12 (referenced by NM_012338 for mRNA) were validated and selected as candidate variants for further analysis.
Sanger sequencing was employed to validate the suspected variants identified within DNA segments of both probands and their family members, ensuring the verification of exon sequencing results. Specific primers tailored to encompass the potential variant sites within LRP5 and TSPAN12 were designed utilizing the Premier 5 software (PREMIER Biosoft, USA). These primers facilitated the amplification of DNA fragments harboring the pathogenic variant sites. Following amplification, these DNA fragments were incorporated into a PCR mixture comprising DNA primers, DNA polymerase, deoxyribonucleotide triphosphates (dNTPs, essential for DNA synthesis), and an appropriate buffer solution. The ensuing PCR process was cyclic, incorporating stages such as DNA denaturation, primer annealing, and DNA elongation. Post-PCR, the amplified products underwent purification via the QIAquick PCR purification kit (QIAGEN, Germany) and were then sequenced using the 3730xl Genetic Analyzer (Applied Biosystems, USA). Sequences derived from the family members were meticulously aligned against the target DNA fragment sequences from the probands. Using Chromas software (Technelysium Pty Ltd, Australia), these sequences, encompassing those of both the probands and their family members, were juxtaposed against a reference sequence, ensuring an accurate determination of the target region's DNA sequence.
Ultimately, co-segregation studies were conducted among the family members [32, 33], and the potential deleteriousness was assessed based on the American College of Medical Genetics and Genomics (ACMG) guidelines [34].
Conservation analysis of LRP5 amino acids
The amino acid sequences of LRP5 proteins from various species were obtained from the universal protein resource (UniProt) [35] and analyzed for conservation among the species using ugene 48.1 software (Biomatters Ltd. New Zealand) for multiple sequence alignment. A phylogenetic tree was constructed to summarize the evolutionary and phylogenetic relationships among the species.
The three-dimensional structure modeling
The three-dimensional (3D) structures of both the wildtype and mutations of LRP5 were generated using Alphafold2 [36] based on the amino acid sequence obtained from the UniProt website through multiple sequence alignment. The protein models were scored using the frame-aligned point error function (FAPE) [37], which is based on neural network training. The top-ranked result was selected to provide the structure for dynamic simulation, and the protein structure was visualized using PyMol 2.5.32 software [38]. The quality of the designed structure of the three proteins was evaluated using Ramachandran plots [39].
Molecular dynamic simulation
To investigate the stability and conformational changes of the entire protein at the atomic level, we conducted a 200 ns MD simulation for both wildtype and mutant structures using the GROMACS 2019.6 software package (Department of Chemistry and Biotechnology, Sweden). The Amber14sb all-atom force field was chosen for MD simulations based on its extensive parameterization and its exceptional suitability for simulating complex macromolecular systems, including membrane proteins like LRP5 [40]. The wildtype and mutant protein structures underwent solvation in a transferable interatomic potential with three points (TIP3P) water model, in a cubic water box with a separation of 1 Å between the protein structure and the box edges, using the periodic boundary conditions to avoid edge effects [41, 42]. The wildtype system required 98 Na + ions for neutralization, compared to 58 for p.Trp691Cys and 55 for p.Pro1431Argfs*8 mutants. Then relaxed by the steepest descent algorithm and the conjugate gradient algorithm within 50,000 steps. The coulomb force and van der Waals interactions were calculated based on a cut-off distance of 1.4 nm. The protein topology was established following the aforementioned steps and then equilibrated using the NVT (isochoric-isothermal) and NPT (isothermal-isobaric) ensembles [43]. Subsequently, a 200 ns MD simulation was conducted, during which data on root mean square deviation (RMSD), root mean square fluctuation (RMSF), and radius of gyration (RG) were collected for subsequent analysis. Hydrogen bond number (HBNUM) and secondary structure calculations were carried out by the dictionary secondary structure pattern (DSSP) algorithm [44]. Snapshots of the system were captured at regular intervals of 50 picoseconds for subsequent analyses. Trajectories of the C-α atoms were plotted employing Origin 85 software (OriginLab Corporation, USA). Furthermore, to elucidate the dynamic conformational changes in the three proteins, we generated graphical representations as well as video clips to visualize the structural variations throughout the entirety of the MD simulation. These visualizations were performed using PyMOL 2.5.32 software platforms. Accompanying video materials are provided in Additional File 1.
Molecular docking
DKK1 is a pivotal protein involved in the regulation of the Wnt signaling pathway. As a member of the Dickkopf protein family, it acts as a Wnt antagonist. DKK1 exerts its function by binding to the LRP5 co-receptors, thereby impeding Wnt signaling activation and suppressing the canonical Wnt pathway [45]. The AlphaFold-predicted 3D model of DKK1 was acquired from the UniProt website and subsequently processed using PyMOL version 2.5.32. This processing involved the removal of water molecules, hydrogen atoms, and extraneous protein regions.
In the docking experiments, ZDOCK version 3.0.2 [46] served as the computational tool for executing global rigid docking. The crystal structure, identified by PDB ID 5GJE, provided a reference framework for establishing the interaction interface between DKK1 and LRP5. Specifically, the C-terminal region of DKK1 (residues 178–266) was anchored to the highly conserved third and fourth YWTD-EGF-β-propeller domains (residues 631–1246) of LRP5. Subsequent docking involved aligning the N-terminal region of DKK1 with the first and second YWTD-EGF-β-propeller domains of LRP5.
Upon completing the docking, a 30 ns MD simulation was conducted using GROMACS 2019.6 and the Amber14sb all-atom force field, by the procedures detailed in the "Molecular Dynamics Simulation" section. Subsequently, binding free energy was calculated employing the MM/GBSA method [47, 48]. Trajectory snapshots were saved at 10 ps intervals for clustering analysis. The structure with the highest clustering frequency was then scrutinized for binding modes via PyMOL version 2.5.3.
Alanine scanning mutagenesis
Alanine scanning is a widely used technique for investigating the role of specific amino acid residues in a protein's function. By replacing these residues with alanine, which has minimal structural impact due to its small, nonpolar nature, the technique allows for an assessment of the consequences of missense variants. It also contributes to our understanding of the functional significance of each residue [49]. To analyze the local residue changes and binding free energies resulting from alanine mutations, we employed alanine scanning by manually introducing alanine mutations at positions 691 and 1431 of the wildtype LRP5 protein. Subsequently, we conducted 30 ns MD simulations and calculated binding free energies for the complex between the alanine mutant protein and DKK1, using the methodology described in the docking section.
Statistical analysis
The comparison between the wildtype and two mutant proteins (p.Trp691Cys and p.Pro1431Argfs*8) was performed using Tukey's method with SPSS 26.0 software (IBM Corporation, USA). Statistical significance was determined based on a threshold P-value of less than 0.05.]
To elaborate on the methodology, we created a flowchart to illustrate the experimental process, see Fig. 1.

The flowchart illustrates the sequencing and computer simulation processes
Results
Clinical manifestations
The general data, visual acuity results, fundus photographs, and FFA of eight affected individuals, including the proband and first-degree relatives from four families were shown in Table 1. The probands consisted of three males and one female, with a mean age of 12.25 years (range, 7 to 17 years), while the affected family members included one male and three females, with a mean age of 43.25 years (range, 42 to 45 years). BCVA ranged from finger count before the eye to 1.0. Notably, fundus manifestations varied significantly among individuals, even within the same family. 3 out of 4 probands were categorized as stage 3, while their parents were classified as stage 2. Specifically, the FFA of the probands from three families exhibited RD in either of the eyes, except for the proband from family 1, whose fundus only showed dilated blood vessels and fluorescein leakage. In contrast, their parents consistently showed no perfusion area, increased vascular branching, and stained vascular walls (Fig. 2).

Fundus Photography and Fluorescein Angiography of Individuals Affected by FVER. Fundus photographs and fluorescein angiography images of probands in four different families are presented as increased vascular branches, fluorescein leakage, no perfusion area scleral buckling or paraoptic disc retinal atrophy, retinal detachment, while their parents only showed no perfusion area and increased vascular branches or fluorescein leakage. Abbreviations: FVER stands for familial exudative vitreoretinopathy, OD for the right eye, and OS for the left eye
Variants detected by targeted genetic sequencing
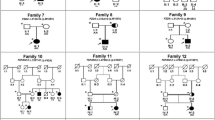
Four variants were detected in 4 out of 9 (44%) families (Table 2, Fig. 3A). Among these, two novel heterozygous variants (c.4289delC: p.Pro1431Argfs*8 and c.2073G > T: p.Trp691Cys) were identified in LRP5, while the other two variants (c.1801G > A: p.Gly601Arg in LRP5 and c.633 T > A: p.Tyr211* in TSPAN12) had been previously reported [50, 51].

Genetic findings in four families and conservative analysis of a novel missense variant. A Pedigrees of the families with causative variants and the corresponding Sanger sequencing results of a heterozygous variant in the proband and his/her first-degree non-affected relative. Squares represent males, circles represent females, arrows indicate the proband, open symbols represent unaffected individuals, solid symbols represent affected individuals, ± represents heterozygous variant, and + / + represents wildtype. B Alignment of LRP5 homologous protein sequences from 26 species, using human LRP5 as a reference, reveals a remarkable conservation of the tryptophan residue at position 691 across these species. C The phylogenetic trees represent the evolutionary relationships of various species
The newly identified variant c.4289delC: p.Pro1431Argfs*8 in LRP5 caused a Pro to Arg substitution at position 1431, followed by premature termination at position 1437 due to eight frameshift amino acids. This resulted in a truncated protein product. The second novel variant, c.2073G > T: p.Trp691Cys, was found in family 2 and led to a Trp to Cys substitution. These two variants were absent in other healthy family members but were detected in the probands and affected relatives.
Two variants, c.1801G > A: p.Gly601Arg in LRP5 and c.633 T > A: p.Tyr211* in TSPAN12, were identified in families 3 and 4, respectively, and have been previously reported. The c.1801G > A missense variant in LRP5 resulted in the substitution of Gly with Arg at position 601, while the c.633 T > A missense variant in TSPAN12 led to a nonsense mutation at Tyr position 211, resulting in protein truncation.
Pathogenicity analysis
While sequencing validation was not conducted in the control population, the two variants in the LRP5 gene (c.4289delC: p.Pro1431Argfs*8 and c.2073G > T: p.Trp691Cys), as well as one variant in the TSPAN12 gene (c.633 T > A: p.Tyr211), were not identified in public databases such as dbSNP, ExAC, or 1000 Genomes. In contrast, the minimum allele frequency (MAF) of the c.1801G > A: p.Gly601Arg variant was 0.0000083.
In silico analysis methods SIFT, Polyphen-2, REVEL (scored 0.994, and 0.965 respectively), MutationTaster, and GERP + + (scored 4.11, and 4.13 respectively) predicted two of the variants (c.2073G > T: p.Trp691Cys and c.1801G > A: p.Gly601Arg) to be disease-causing or damaging. The remaining two variants (c.4289delC: p.Pro1431Argfs*8 and c.633 T > A: p.Tyr211*) were predicted to be disease-causing or damaging by MutationTaster and GERP + + (scored 4.53 and 5.68 respectively) (Table 2). All four variants exhibited co-segregation among affected family members. Notably, the newly identified missense variant c.2073G > T: p.Trp691Cys is evolutionarily conserved across a broad spectrum of species, extending from humans to zebrafish (Fig. 3B and C). Therefore, all these variations were considered pathogenic according to the ACMG standard.
Protein structure evaluation
The Ramachandran plot analysis of the three structures in Fig. 4 reveals that amino acid residues falling within the favored regions make up more than 90% of the entire protein. This suggests that the models' conformations conform to the fundamental principles of stereochemistry [52].

The protein structures were modeled using AlphaFold2 and their conformations were evaluated via Ramachandran plots. A For the wildtype protein, 91.57% of amino acid residues were found in the favored regions. B In the p.Trp691Cys mutant, 90.08% of residues were in the favored regions. C The p.Pro1431Argfs*8 mutant showed 93.87% of its residues in the favored areas
Molecular dynamic simulation and data analysis
RMSD and RMSF have commonly used measures to assess the spatial variations of proteins in MD simulations. RMSD quantifies the overall difference of a molecule compared to a reference conformation. The RMSD values of the wildtype and two LRP5 mutations were illustrated in Fig. 5A. During the simulation, the p.Trp691Cys variant showed smaller conformational changes than the wildtype (P = 0.000, < 0.05, 95%CI) (Fig. 5E), while the p.Pro1431Argfs*8 variant did not show statistically significant differences in conformational changes compared to the wildtype (P > 0.05, 95% CI). In the end, the backbone atoms of all three proteins remained around 1.5 nm.

RMSD, RMSF, Rg, HBNUM values comparisons between wildtype and two mutant proteins in MD simulation. A-D Evolution over time to show RMSD, RMSF, Rg, and HBNUM values of the wildtype (black) and the two mutant proteins (p.Trp691Cys in red, p.Pro1431Argfs *8 in blue). E Statistical analysis comparison of RMSD and RG values as bar graphs, depicting values with p < 0.05 (95% CI)
The RMSF analysis revealed distinct flexibility profiles among the wildtype and mutant proteins at specific residues (Fig. 5B) The wildtype's most flexible regions were Trp10-Ala27, Asp1318-Ile1387, Met1513-Asn1520, and Pro1606-Ser1615. In contrast, p.Trp691Cys displayed limited flexibility at Leu14-15, Asp1331-1333, and Ser1486-Thr1489; p.Pro1431Argfs8 had flexibility at Met1-Pro28 and Pro1261-Ser1385. Average RMSF values for the active sites fluctuated between 1.03–1.38 nm, 1.02–1.08 nm, and 1.42–1.53 nm for the wildtype, p.Trp691Cys, and p.Pro1431Argfs8, respectively. Maximum RMSF values were 1.87 nm, 1.16 nm, and 2.68 nm, respectively, implying reduced conformational changes in p.Trp691Cys and heightened local activity in p.Pro1431Argfs8, especially at Met1-Pro28 and Pro1261-Ser1385 (Fig. 6A). These findings suggest that the pathogenic variants may influence the flexibility of the protein backbone, which could affect the protein's structure and function. Especially evident in the videos of Additional file 1, the conformational changes of the three proteins over time were well-illustrated. Although the C-terminal region of the p.Pro1431Argfs*8 mutant exhibited heightened activity, it too reached a state of convergence by the end of the simulation.

Comparison of active sites and H-bonds between the wildtype and mutant proteins. A. Changes in active sites in wildtype and two mutant proteins during simulation were marked in red. B. The snapshots extracted from the last frame of the MD simulation showed the intramolecular H-bonds changes in wildtype and mutants. The target residues along with H-bonds were marked in bright green
Rg values, which measure the distance or fluctuation from C-α atoms to the center of mass, were 4.34 nm, 4.25 nm, and 4.45 nm for the wildtype and two mutants, respectively (Fig. 5C). The C-α atoms in the p.Pro1431Argfs*8 mutant showed greater fluctuation (P = 0.000, < 0.05, 95%CI)) compared to the wildtype, while the p.Trp691Cys mutant exhibited less fluctuation (P = 0.000, < 0.05, 95% CI) (Fig. 5E). These results suggest that the p.Trp691Cys variant may increase the rigidity of the protein, whereas the p.Pro1431Argfs*8 variant may enhance its flexibility at the local conformation, especially in the regions of Met1-Pro28 and Pro1261-Ser1385.
HBNUM stands for the number of hydrogen bonds (H-bond), which are crucial interactions for stabilizing the spatial arrangement of proteins. It is evident from the results that the number of H-bonds in p.Trp691Cys is similar to that of the wildtype, while in p.Pro1431Argfs*8, which is a truncated protein due to the absence of C-terminus structures, the overall number of H-bonds is reduced compared to the wildtype (Fig. 5D). In terms of intramolecular interactions through H-bonds, as shown in Fig. 6B, Trp691 formed H-bonds with Ser700 in the wildtype, while the Cys691 of p.Trp691Cys formed H-bonds with Ala677, Ala679, and Ser700. In the p.Pro1431Argfs*8 mutant, the Pro1431 was transformed into Arg, which formed H-bonds with Tyr719, Glu721, and Thr737. The changes in H-bond distribution certainly contributed to the structural and conformational alterations.
To perform DSSP analysis, the last 50 ps of the simulation were selected, and the final frames of the MD simulations for the three proteins were obtained and compared with their respective initial structures. Additionally, a comparison was made between the 3D structures of the wildtype and the two mutant proteins in the final frame. All three ensembles were observed to converge to more stable conformations relative to their initial states, as depicted in Fig. 7A. In the final frame, both mutant proteins exhibited greater structural convergence and rigidity compared to the wildtype protein (Fig. 7C).

Molecular Trajectories and Structural Comparisons of Wildtype and Mutant Proteins in MD Simulation. A Snapshots of molecular trajectories for the three proteins at both the initial and final frames of the simulation are superimposed and color-coded, with the initial frame marked in green and the final frame in red. B Quantitative analysis of eight principal secondary structures (coil, β-sheet, β-bridge, bend, turn, α-helix, 5-helix, and 3-helix) in the wildtype and the two mutants, p.Trp691Cys and p.Pro1431Argfs*8. C The figure displays snapshots of molecular trajectories at the final frame of the simulation, comparing the wildtype and two mutant proteins. The wildtype protein is marked in grey, while the two mutant proteins are denoted in green
Figure 7B presents the DSSP analysis results. Post-simulation, the wildtype protein showed a decrease in coil, β-sheet, and turn structures, while witnessing an increase in bend and 3-helix formations. The α-helix configuration remained constant, and a β-bridge structure emerged. Importantly, the 5-helix structure was absent throughout the entire simulation period.
Before the simulation, p.Trp691Cys had fewer β-sheet and turn structures than the wildtype but more bend, 3-helix, and α-helix structures. Post-simulation, turn and α-helix structures increased, surpassing the wildtype, while β-sheet remained stable. The β-bridge disappeared, and no 5-helix was observed at any stage.
For p.Pro1431Argfs*8, pre-simulation levels of β-sheet were similar to the wildtype, but turn structures were less abundant. Bend, 3-helix, and α-helix were more prevalent. Post-simulation, turn structures increased to wildtype levels; bend and α-helix decreased while 3-helix rose, still lagging behind the wildtype. A 5-helix structure appeared post-simulation. Throughout, the coil structure was consistently less abundant than the wildtype. The DSSP analysis is visually represented in Additional File 2.
Molecular interactions
In the docking simulation, the binding energies for wildtype-DKK1, p.Trp691Cys-DKK1, and p.Pro1431Argfs*8-DKK1 were -90.37 ± 2.35 kcal/mol, -135.78 ± 2.75 kcal/mol, and -118.69 ± 5.38 kcal/mol, respectively. Residue decomposition showed that hydrophobic and electrostatic interactions mainly drove binding, followed by nonpolar solvation-free energy. Both mutant proteins displayed larger negative values for van der Waals and electrostatic energies compared to the wildtype, indicating a stronger affinity for DKK1. Solvation factors for the mutants also suggested a more favorable binding environment with DKK1 (Table 3). These results indicate that both p.Trp691Cys-DKK1 and p.Pro1431Argfs*8-DKK1 have higher binding affinity compared to wildtype-DKK1 protein. Therefore, the pathogenic variants enhance the binding propensity of the protein with DKK1.
Figure 8 illustrates the H-bond interactions between DKK1-C (residues 178–266) and the conserved third and fourth YWTD-EGF-β-propeller domains (residues 631–1246) of LRP5. Specifically, nine residues on DKK1 form H-bonds with nine residues on the wildtype protein.

The 3D binding modes of the wildtype, p.Trp691Cys, and p.Pro1431Argfs*8 systems with DKK1 protein are shown, with a focus on the docking site. The LRP5 protein is represented in purple, while the DKK1 protein is depicted in a yellow–brown color
In the p.Trp691Cys-DKK1 complex, 11 residues from each protein are involved in H-bond formation. Similarly, in the p.Pro1431Argfs*8-DKK1 complex, 10 residues from each protein contribute to hydrogen bonding. These interactions provide insight into the differential affinities and stabilizing factors in the binding of wildtype and mutant LRP5 to DKK1.
Compared to the wildtype, both the p.Trp691Cys and p.Pro1431Argfs*8 mutants exhibit an increased number of H-bonds in their protein binding complexes, along with a change in the residue types that interact with DKK1 protein. Details on the specific H-bond interactions and bond lengths can be found in Additional File 3.
Alanine mutagenesis analysis
By comparing the mutated residues with alanine residues, the impact of point mutations on protein structure is elucidated. As shown in Additional File 4, the alanine variants at positions 691 and 1431 lead to a decrease in binding free energies and an increase in binding affinity compared to the wildtype. The binding free energy for the p.Trp691Ala is -134.75 ± 3.96 kcal/mol, while the binding free energy for the p.Pro1431Ala is -95.54 ± 3.18 kcal/mol. In contrast, the wildtype binding free energy is -90.37 ± 2.35 kcal/mol.
Discussion
In this study, 3 variants of LRP5 and 1 of TSPAN12 in four unrelated FEVR families were found to be pathogenic according to the ACMG guidelines. Two of the LRP5 variants, c.4289delC (p.Pro1431Argfs*8) and c.2073G > T (p.Trp691Cys), were novel variants with extensive changes in protein structure according to MD simulation. While the other two variants, c.1801G > A (p.Gly601Arg) in LRP5 and c.633 T > A (p.Tyr211*) in TSPAN12, have been previously reported [50, 51]. It is reported that approximately 50% of FEVR patients harbored deleterious variants [3], among which LRP5 accounts for about 12%—25% and TSPAN12 for about 3%—10% [8, 53,54,55,56], and that the mutation frequency of the same gene varies in different ethnicities. In this study, the total detection rate of all variants was 44% (4/9), which was consistent with previous reports [3]. Of all the FEVR patients, 75% (3/4) were caused by the LRP5 variant, whereas 25% (1/4) were by the TSPAN12 variant. However, Zou. et, al. [51] observed that in the same region of China (Ningxia), four out of eight probands (50%, 4/8) harbored pathogenic variants in TSPAN12 (75%, 3/4) and FZD4 (25%, 1/4) genes, but none of them carried LRP5 variants. Combining the data from the two research revealed that in Ningxia, LRP5 accounted for 37.5% (3/8) and TSPAN12 for 50% (4/8) of FEVR patients. As a result, the LRP5 and TSPAN12 genes were therefore hypothesized to be the more prevalent causative genes in the Ningxia population. Certainly, it is still necessary to verify these findings with a larger sample size to rule out the possibility of selection bias in subsequent investigations. Due to the absence of any literature indicating gender differences in this disease, we did not investigate gender disparities in our study.
As previously documented, variants in LRP5, exhibiting autosomal dominant, recessive, and X-linked inheritance patterns, lead to disturbances in the Wnt signaling pathway. These disruptions, in turn, result in aberrant retinal vascular development, giving rise to the onset of lesions associated with familial exudative vitreoretinopathy (FEVR) [57, 58]. Additionally, it has been reported that the recessive inheritance pattern of LRP5 not only causes abnormalities in retinal vascular development but also gives rise to severe skeletal anomalies [58]. In this study, we identified three pathogenic variants of LRP5 with an autosomal dominant inheritance pattern, among which the novel missense variant c.2073G > T (p.Trp691Cys) was found. This variant is located in the highly conserved third YWTD-EGF-β-propeller domains. Studies of LRP6 suggest that the third YWTD-EGF repeats are the region to bind with ligands of the Wnt signal pathway [45]. Considering the high similarity between LRP5 and LRP6 proteins [45], It is plausible to hypothesize that the c.2073G > T (p.Trp691Cys) variant in LRP5 could potentially impact the Wnt signaling pathway by influencing the binding of extracellular ligands to the YWTD-EGF-β-propeller domains. Another frameshift variant of LRP5, c.4289delC (p.Pro1431Argfs*8), was first identified in this study, which resulted in a premature termination of the protein at position 1437 and generated a truncated protein that may be pathogenic due to the loss of function.
To elaborate the pathogenic mechanism at the dynamic molecular level, we employed alphafold2 to model the 3D structure of LRP5 for the first time and implemented 200 ns MD simulation to analyze the changes in RMSD, RMSF, RG, HBNUM, and DSSP values in wildtype and mutant proteins. These indicators suggest that the p.Trp691Cys and p.Pro1431Argfs*8 variants may have triggered FEVR manifestation by modifying the overall conformation of the LRP5 protein, potentially affecting its ligand binding. Numerous studies and experiments have established that the flexibility of specific regions in proteins plays a crucial role in opening up ligand-binding sites. This dynamic behavior is intimately linked to a variety of biological functions, including molecular docking, catalytic activity, and the transport of small molecules [59,60,61,62,63,64].
In a 200 ns MD simulation of p.Trp691Cys, we observed that the replacement of Trp691 with Cys691 generated new hydrogen bonds with adjacent residues. This increased the protein's structural stability but reduced its flexibility. Conversely, the p.Pro1431Argfs*8 mutation truncated the protein at the 1437th residue, resulting in fewer secondary structures and a reduced number of hydrogen bonds. Additionally, this variant altered the polarity of specific residues, such as changing the positively charged His1432 to a neutral Thr and replacing Glu1437 with a neutral Ser. These changes likely disrupted existing salt bridges, increasing the dynamism of the region between residues 1261 and 1385, thereby altering its spatial arrangement.
Our molecular docking study reveals the significant influence of H-bond interactions on the binding affinity between proteins and ligands. We found that four mutant proteins (p.Trp691Cys, p.Pro1431Argfs*8, p.Trp691Ala, and p.Pro1431Ala) have lower binding free energies and higher affinity for DKK1 compared to the wildtype. Specifically, the p.Trp691Cys and p.Trp691Ala variants show a markedly reduced binding free energy, emphasizing the importance of the Trp691 residue within the conserved third YWTD-EGF-β-propeller domain in the protein's function and structure. These findings suggest that these mutants could enhance their binding affinity and stability with DKK1, possibly playing a crucial role in modulating the Wnt signaling pathway [45]. The enhanced binding could lead to increased inhibition of LRP5 by DKK1, potentially suppressing Wnt signaling through the promotion of β-catenin degradation.
In our study, we observed that despite sharing the same pathogenic variant, probands and their parents exhibited different clinical phenotypes. Specifically, parents of patients with severe FEVR typically displayed mild manifestations, which is consistent with a previous report by Gilmour [2]. Additionally, probands from different families who harbored the same variant showed varying levels of severity in the disorder. For example, in our study, proband 4 carrying the TSPAN12 variant (c.633 T > A: p.Tyr211*) experienced retinal detachment, while the same variant was detected by Zou et al. [51]. resulted in milder conditions such as vitreous hemorrhage and peripheral avascular area or vascular exudate. Furthermore, a separate study [50] reported that younger FEVR patients tend to display more severe phenotypes than older patients. Gilmour et al. [2] have speculated that individuals carrying the same pathogenic variant often present different phenotypes due to the influence of epigenetics and environmental factors. They found that even minor changes in oxygen content or intrauterine conditions, such as PaO2 alteration or exposure to certain drugs, may have a considerable impact on patients who harbor FEVR pathogenic variants.
Nevertheless, our study does have the following limitations that should be acknowledged. Firstly, the small sample size may potentially introduce sample selection bias and hinder the detection of rare pathogenic variants. Additionally, the limitations of the sequencing technology used may have hindered the detection of novel pathogenic variants or copy number variants related to FEVR. Furthermore, in vitro or animal laboratory testing is needed to confirm the pathogenicity of the two novel variants (c.2073G > T: p.Trp691Cys and c.4289delC: p.Pro1431Argfs*8) of LRP5. To address these limitations, future studies could expand the sample size and include a broader range of family members for TGS. Additionally, integrating laboratory testing would allow for a more comprehensive analysis of genetic variations.
Conclusion
This study successfully identified two new pathogenic variants in the LRP5 gene (c.2073G > T: p.Trp691Cys and c.4289delC: p.Pro1431Argfs*8) associated with FEVR, leveraging both mutation screening and MD simulations. This expands our knowledge of FEVR-related mutations and opens new doors for genetic research in FEVR patients. We strongly advocate for family counseling and genetic education programs to offer comprehensive insights into the disease's genetic aspects for affected individuals and families. Periodic eye screenings are also recommended for early diagnosis and tailored treatment approaches. Emphasizing interdisciplinary collaboration can facilitate knowledge exchange and comprehensive patient care, aiming to enable early diagnosis, improve personalized treatments, and ultimately lower FEVR prevalence.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the [National Library of Medicine] repository, (https://www.ncbi.nlm.nih.gov/sra/PRJNA922952).
References
Criswick VG, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol. 1969;68:578–94.
Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye. 2015;29:1–14.
Kashani AH, Brown KT, Chang E, et al. Diversity of retinal vascular anomalies in patients with familial exudative vitreoretinopathy. Ophthalmology. 2014;121:2220–7.
Ober RR, Bird AC, Hamilton AM, et al. Autosomal dominant exudative vitreoretinopathy. Br J Ophthalmol. 1980;64:112–20.
Xia CH, Liu H, Cheung D, et al. A model for familial exudative vitreoretinopathy caused by LPR5 mutations. Hum Mol Genet. 2008;17:1605–12.
Boonstra FN, van Nouhuys CE, Schuil J, et al. Clinical and molecular evaluation of probands and family members with familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2009;50:4379–85.
Nikopoulos K, Gilissen C, Hoischen A, et al. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86:240–7.
Seo SH, Yu YS, Park SW, et al. Molecular characterization of FZD4, LRP5, and TSPAN12 in familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2015;56:5143–51.
Salvo J, Lyubasyuk V, Xu M, et al. Next-generation sequencing and novel variant determination in a cohort of 92 familial exudative vitreoretinopathy patients. Invest Ophthalmol Vis Sci. 2015;56:1937–46.
Rao FQ, Cai XB, Cheng FF, et al. mutations in LRP5, FZD4, TSPAN12, NDP, ZNF408, or KIF11 genes account for 38.7% of Chinese patients with familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2017;58:2623–9.
Niehrs C, Acebron SP. Mitotic and mitogenic Wnt signaling. Embo J. 2012;31:2705–13.
Zhang S, Li X, Liu W, et al. Whole-Exome Sequencing Identified DLG1 as a Candidate Gene for Familial Exudative Vitreoretinopathy. Genet Test Mol Biomarkers. 2021;25:309–16.
Hildebrand PW, Rose AS, Tiemann JKS, et al. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem Sci. 2019;44:902–13.
Imani S, Cheng J, Shasaltaneh MD. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget. 2017;9:122–41.
Jin X, Zhang J, Yi Q, et al. Leber’s hereditary optic neuropathy arising from the synergy between ND1 3635G>A mutation and mitochondrial YARS2 mutations. Invest Ophthalmol Vis Sci. 2021;62:22.
Preising MN, Görg B, Friedburg C, et al. Biallelic mutation of human SLC6A6 encoding the taurine transporter TAUT is linked to early retinal degeneration. FASEB J. 2019;33:11507–27.
Vocke F, Weisschuh N, Marino V, et al. Dysfunction of cGMP signaling in photoreceptors by a macular dystrophy-related mutation in the calcium sensor GCAP1. Hum Mol Genet. 2016;26:133–44.
Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99:1129–43.
Canny CL, Oliver GL. Fluorescein angiographic findings in familial exudative vitreoretinopathy. Arch Ophthalmol. 1976;94:1114–20.
dbSNP. https://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi. Accessed 10 Apr 2022.
ExAC. https://gnomad.broadinstitute.org/. Accessed 10 Apr 2022.
gnomAD. https://gnomad.broadinstitute.org/. Accessed 10 Apr 2022.
1000genomes. https://www.internationalgenome.org/. Accessed 10 Apr 2022.
SIFT. https://sur.ly/i/sift.jcvi.org/. Accessed 10 Apr 2022
MutationTaster. https://www.mutationtaster.org/. Accessed 10 Apr 2022
PolyPhen-2. http://genetics.bwh.harvard.edu/pph2/. Accessed 10 Apr 2022
REVEL. https://ngdc.cncb.ac.cn/biocode/tools/BT004461/stat. Accessed 10 Apr 2022
GERP++. http://mendel.stanford.edu/sidowlab/downloads/gerp/index.html. Accessed 10 Apr 2022
Jin ZB, Li Z, Liu Z, et al. Identification of de novo germline mutations and causal genes for sporadic diseases using trio-based whole-exome/genome sequencing. Biol Rev Camb Philos Soc. 2018;93:1014–31.
ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/. Accessed 10 Apr 2022
HGMD. https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database/. Accessed 10 Apr 2022.
Jin ZB, Ito S, Saito Y, et al. Clinical and molecular findings in three Japanese patients with crystalline retinopathy. Jpn J Ophthalmol. 2006;50:426–31.
Xing DJ, Zhang HX, Huang N, et al. Comprehensive molecular diagnosis of Bardet-Biedl syndrome by high-throughput targeted exome sequencing. PLoS ONE. 2014;9:e90599.
Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19:1105–17.
UniProt. https://www.uniprot.org/. Accessed 21 May 2022.
Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:439–44. https://alphafold.ebi.ac.uk. Accessed 23 May 2022.
Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9.
Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–91 (https://pymol.org/2/).
Structure Assessment - Expasy. https://swissmodel.expasy.org/assess. Accessed 23 May 2022.
Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–713.
Van Gunsteren WF, Berendsen HJ, Hermans J, et al. Computer simulation of the dynamics of hydrated protein crystals and its comparison with x-ray data. Proc Natl Acad Sci USA. 1983;80:4315–9.
Cheatham TE, Miller JL, Fox T, et al. Molecular dynamics simulations on solvated biomolecular systems: the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J Am Chem Soc. 1995;117:4193–4.
Hess B, Bekker H, Berendsen HJC, et al. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–72.
Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–637.
Mao B, Wu W, Li Y, et al. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411:321–5.
Pierce BG, Hourai Y, Weng Z, et al. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE. 2011;6:e24657.
Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10:449–61.
Rastelli G, Rio AD, Degliesposti G, et al. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem. 2010;31:797–810.
Oyarzún DA, et al. Alanine scanning mutagenesis: a practical approach for exploring protein function. Methods Mol Biol. 2013;987:219–34.
Li JK, Li Y, Zhang X, et al. Spectrum of variants in 389 Chinese probands with familial exudative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2018;59:5368–81.
Zou G, Qi R, Ma MJ, et al. A novel stop codon mutation of TSPAN12 gene in Chinese patients with familial exudative vitreoretinopathy. Ophthalmic Genet. 2022;43:210–7.
Laskowski RA, MacArthur MW, Moss DS, et al. PROCHECK - a program to check the stereochemical quality of protein structures. J App Cryst. 1993;26:283–91.
Kondo H, Kusaka S, Yoshinaga A, et al. Mutations in the TSPAN12 gene in Japanese patients with familial exudative vitreoretinopathy. Am J Ophthalmol. 2011;151:1095–100.
Yang H, Xiao X, Li S, et al. Novel TSPAN12 mutations in patients with familial exudative vitreoretinopathy and their associated phenotypes. Mol Vis. 2011;17:1128–35.
Qin M, Hayashi H, Oshima K, et al. Complexity of the genotype-phenotype correlation in familial exudative vitreoretinopathy with mutations in the LRP5 and/or FZD4 genes. Hum Mutat. 2005;26:104–12.
Yang H, Li S, Xiao X, et al. Identification of FZD4 and LRP5 mutations in 11 of 49 families with familial exudative vitreoretinopathy. Mol Vis. 2012;18:2438–46.
Ye X, Wang Y, Cahill H, et al. Norrin, Frizzled-4, and LRP5 Signaling in Endothelial Cells Controls a Genetic Program for Retinal Vascularization. Cell. 2009;139:285–98.
Downey LM, Bottomley HM, Sheridan E, et al. Reduced bone mineral density and hyaloid vasculature remnants in a consanguineous recessive FEVR family with a mutation in LRP5. Br J Ophthalmol. 2006;90:1163–7.
Kay LE. NMR studies of protein structure and dynamics. J Magn Reson. 2005;173:193–207.
Palmer AG. NMR characterization of the dynamics of biomacromolecules. Chemical Rev. 2004;104:3623–40.
Yap KL, Yuan T, Mal TK, et al. Structural basis for simultaneous binding of two carboxy-terminal peptides of plant glutamate decarboxylase to calmodulin. J Mol Biol. 2003;328:193–204.
Schumacher MA, Rivard AF, Bächinger HP, et al. Structural of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–4.
Schlessinger A, Rost B. Protein Flexibility and Rigidity Predicted From Sequence. Proteins. 2005;61:115–26.
King AE, Ackley MA, Cass CE, et al. Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharmacol Science. 2006;27:416–25.
Acknowledgements
We would like to express our sincere gratitude to all the patients and their family members who participated in this study. Their willingness to contribute and participate in the research has been invaluable. We would also like to acknowledge the generous support provided by the funding agencies, including the National Natural Science Foundation of China (82060182), Ningxia Natural Science Foundation (2021AAC03301), and the Fundamental Research Funds for the Central Universities (31920200027). Their financial support has played a crucial role in the successful completion of this study.
Funding
The National Natural Science Foundation of China (82060182), Ningxia Natural Science Foundation (2021AAC03301), and the Fundamental Research Funds for the Central Universities (31920200027) have provided funding support for gene sequencing and MD simulation.
Author information
Authors and Affiliations
Contributions
Li JY and Wang CJ collected the data and wrote the main manuscript. Zhang SH, Cai B, Pan B, Sun CH, Qi XL, Ma CM, Fang W and Jin KX analyzed and interpreted the patient data regarding the FEVR disease. Bi XJ, Jin ZB and Zhuang WJ designed the work and substantively revised the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was performed by the Declaration of Helsinki and approved by the Ethics Committee of Ningxia Hui Autonomous Region People Hospital (No. 2020-KY-GZR019). The informed consent was signed by each participant or the guardians in the study.
Consent for publication
The participants included in this research manuscript have provided written informed consent to have their data and/or images published. They were fully informed about the purpose, nature, and potential consequences of publishing their information in the manuscript. All personal identifiers have been removed or disguised to ensure anonymity and privacy. The participants understood that their participation was voluntary and that they had the right to withdraw their consent at any time without any repercussions. The authors of this manuscript take full responsibility for protecting the confidentiality and privacy of the participant's data.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The trajectory animations depict the dynamic behavior of a wildtype protein and two mutant proteins (p.Trp691Cys and p.Pro1431Argfs8) during the molecular dynamics simulation.
Additional file 2.
Visual Representation of DSSP Analysis During the course of a 200ns molecular dynamics simulation, discernible alterations were observed in the secondary structural elements of the wild-type, p.Trp691Cys and p.Pro1431Argfs*8 proteins. The diagram employs a color-coded scheme to facilitate easy interpretation of these secondary structures: White denotes 'coil'; Red indicates 'β-sheet'; Black signifies 'β-bridge'; Green marks 'bend'; Yellow highlights 'turn'; Blue stands for 'α-helix'; Purple designates '5-helix'; and Gray corresponds to '3-helix'.
Additional file 3.
Comparisons of the hydrogen bond interactions and distance between wildtype-DKK1, p.Trp691Cys-DKK1, and p.Pro1431Argfs*8-DKK1 during molecular docking.
Additional file 4.
Comparisons of the molecular docking results of the alanine mutagenesis study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, J., Wang, C., Zhang, S. et al. Genetic detection of two novel LRP5 pathogenic variants in patients with familial exudative vitreoretinopathy. BMC Ophthalmol 23, 489 (2023). https://doi.org/10.1186/s12886-023-03243-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-023-03243-2