
Abstract
Background
Pediatric Behçet’s disease (PBD) is rarer than BD and can be a challenging diagnosis as clinical picture may be incomplete. As in adult patients, sight-threatening ocular manifestations may lead to diagnosis. In this study, we aimed to report a series of cases of PBD with ocular manifestations and provide a review of the literature.
Methods
Retrospective case series of PBD patients with ocular manifestations. Demographic, ophthalmological and systemic data at presentation and during follow-up were collected and analyzed.
Results
Four patients, aged 13.0 ± 2.9 years (9–16) were included. Posterior uveitis with retinal vasculitis, papillitis and macular edema was present in all patients, with associated anterior uveitis in 2 cases. Other features included occlusive vasculitis (2/4) and necrotizing retinitis (2/4). All patients were improved by systemic treatments except one patient with severe bilateral optic neuropathy. Ocular manifestations were the presenting symptoms in 3/4 cases.
Conclusion
Ocular manifestations and systemic associations of PBD are comparable to those encountered in adult patients. The lack of complains in pediatric patients may lead to a longer diagnosis delay, especially in unilateral uveitis. Aggressive and long-term treatment is mandatory to prevent vision loss and recurrences.
Similar content being viewed by others
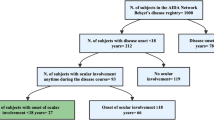


Background
Behcet’s disease (BD) is an immune-mediatedvasculitis involving vessels of all sizes, which presents with multisystemic features and sight-threatening ocular manifestations [1].The course of the disease may be recurrent and refractory, requiring chronic treatments with potential adverse effects [1].
There is a high geographic variation in the incidence rate of BD: most of the countries with a high incidence (20 to 420/100.000) are located along the historical silk road, Turkey having the highest rate in the world [1, 2]. While the peak age of onset is in the young adult population (20–30 years old), onset of BD can occur in children (before 16 years) in 3% to 26% of cases [3,4,5]. In pediatric Behçet’s disease (PBD), the diagnosis may represent a clinical challenge as it commonly features an incomplete clinical picture [6]. Thus, a definition of PBD was recently proposed by the Consensus classification criteria for pediatric Behçet’s disease study (PEDBD) to take into account these peculiarities [7]. The following typical ocular involvements are included in the diagnostic criteria: anterior uveitis, posterior uveitis and retinal vasculitis [7]. These diagnostic criteria include ophthalmological, neurological and vascular items, but oral aphthosis is not mandatory [7]. As PBD is a rare condition, it is not clear whether ocular manifestations are really different from those encountered in adult patients [8]. We here report a case series of PBD with ocular manifestations and provide a review of the literature about this rare but severe clinical situation.
Methods
We retrospectively included pediatric patients (Age ≤ 18 y.o. at diagnosis) with Behçet related uveitis, managed at the Department of Ophthalmology in Bicêtre hospital, France, between 2012 and 2020.
BD diagnosis was made using the PEDBD criteria. If the patient did not meet PEDBD criteria, diagnosis could be made using ICBD (International criteria for Behçet’s disease study [9]) criteria or after multidisciplinary agreement. PEDBD and ICBD criteria are available in Table 1. Medical files were used to analyze demographic data (age, ethnicity, medical history), and extra-ocular involvement. Ophthalmological data included initial and final best-corrected visual acuity (BCVA), uveitis classification according to the Standardization of Uveitis Nomenclature (SUN) classification criteria [10] slit lamp and fundus features. Fluorescein angiography and macular optical coherence tomography (OCT) findings were analyzed. Systemic and topical treatments, clinical response, and long-term follow-up were analyzed for each patient. The study was performed according to regulations of the local ethics committee. Institutional Review Board (IRB)/Ethics Committee approval was obtained by the French Society of Ophthalmology (IRB 00008855 Société Française d’Ophtalmologie IRB#1).
Results
Four patients (3 males, 1 female) aged 13.0 ± 2.9 years (9–16) were included. They were all from North African descent. One patient had a heterozygous mutation of the familial Mediterranean fever gene (MEFV). Oral aphthosis was present in 3 patients, while none had both oral and genital aphtosis. Neurological manifestations were present in 3 cases. Ocular involvement led to the diagnosis of BD in all cases. Three patients met the PEDBD criteria, while 1 patient with typical neurological and ocular involvement was diagnosed as BD after multidisciplinary agreement. Three patients met the ICBD criteria. Presenting ocular symptoms were red eye (2/4) and loss of vision (4/4). Posterior uveitis with retinal vasculitis and papillitis was present in all patients, macular edema was present in 2 cases. Associated anterior uveitis was present in 2 cases. Other features included occlusive vasculitis (2/4), and necrotizing retinitis (2/4). Corticosteroid combined with azathioprine and anti-TNFα were administered in all patients (doses available in Table 2). One patient with severe bilateral optic neuropathy also received cyclophosphamide pulses. He had complete bilateral vision loss caused by optic atrophy during follow-up. Among the 3 remaining, BCVA of the most severe eye improved from 1.5 ± 1.1 (0.2–2.3) at presentation to 0.0 ± 0.1 LogMAR (0–0.1) at the last visit. Mean delay before diagnosis was 11.3 ± 8.5 months (0.3–21). No patient relapsed during follow-up, 63.5 ± 28,6 months (19–104). Detailed results are provided in Table 2.
Case 1
A 13-year-old boy, from Moroccan descent, was referred for blurred vision on the left eye. He was heterozygous for the familial Mediterranean fever gene (M694I), and HLAB-51-positive. He suffered from recurrent fever and oral aphtosis which was diagnosed as PFAPA (periodic fever, aphthous stomatitis, pharyngitis, adenitis) syndrome from his third to his 7th year and was treated with colchicine. He was then lost to follow-up but reported having had recurrent oral ulcers from the age of 7 without genital involvement for which he had never sought medical attention. At the age of 13 he consulted for ocular pain, redness, and sudden drop in visual acuity of his left eye. He reported 3 previous episodes of red eye and floaters which were treated as conjunctivitis at the ages of 11 and 12. He also reported episodes of pseudo-folliculitis and headaches 1–2 times in the preceding 2 years.
BCVA was 20/20 in the right eye and counting fingers on the left eye. Slit lamp examination and intraocular pressure were normal OU. Left fundus examination showed a dense vitreous haze associated with optic nerve and macular edema, without peripheral retinal necrosis, while OD fundus disclosed a mild vitritis. Fluorescein angiogram showed bilateral diffuse capillaropathy, papillitis and peripheral occlusive vasculitis (Fig. 1A, B). Macular OCT confirmed massive macula infiltrate and serous retinal detachment on the left eye (Fig. 1C, D). Brain magnetic resonance imaging (MRI) and lumbar puncture were normal. PBD was diagnosed and the patient was treated with intravenous pulses of methylprednisolone, followed by infliximab tapering oral prednisone associated with colchicine and azathioprine. Macular edema and serous retinal detachment completely resolved (Fig. 1E–H). Argon laser retinal photocoagulation was performed on non-perfused areas. Ocular inflammation was controlled after 3 months with this treatment regimen allowing a gradual decrease in treatment over a total of 4 years (cessation of corticosteroids and infliximab). At the last follow-up (69 months after onset) he had 20/20 OU and was still receiving colchicine and azathioprine.

case #1. A-D Initial visit. A, B Late fluorescein angiogram showing bilateral diffuse capillaropathy and left eye vasculitis. Macular OCT shows preserved right foveal profile (C) while there is a massive macular edema on the left eye with retina infiltrate (D). E–H Last visit. Normalization of the right fluorescein angiogram (E), and inferotemporal pigmented epithelial atrophy in the previous zone of retinitis (F). Note the laser treated peripheral hole in the superotemporal periphery. Normalization of the macular OCT (G, H)
Case 2
A 9-year-old boy, from Tunisian descent, with a history of recurrent oral aphthosis, presented with a rapidly progressive bilateral visual loss with headache.
BCVA was 20/50 OD and counting fingers OS, IOP was normal OU. Biomicroscopic examination showed a massive anterior chamber reaction OU, with posterior synechiae and cyclitic membranes. A dense vitreous haze masked the details of fundus but papillary edema and retinal hemorrhages could be observed. Macular OCT was normal, while fluorescein angiography showed a venous non-occlusive vasculitis associated with diffuse peripheral capillaropathy and optic nerve leakage on both eyes.
Brain MRI was normal and lumbar puncture found an aseptic meningitis with clear aspect, normal glucose and proteins levels, but 66 cells/mm3 with predominance of neutrophils. Polymerase Chain Reaction (PCR) for herpes viruses and bacterial culture were negative. He fulfilled PEDBD criteria with oral aphthosis, ocular and neurological features.
The patient was treated with intravenous Methylprednisolone MP pulses and infliximab, followed with tapering oral prednisone associated with colchicine and azathioprine. Vitreous haze and vasculitis improved rapidly. Twelve months after onset, he recovered normal vision on the right eye, and 20/25 on his left eye, while he was still receiving prednisone, azathioprine, and colchicine. A mild papillary leakage persisted on left fluorescein angiogram.
Case 3
A 14-year-old girl, from Moroccan descent had a history of encephalitis and myelitis two years before ophthalmic presentation, which was treated by intravenous MP pulses. She presented in Ophthalmology with a progressive vision loss of the right eye. BCVA was 20/32 OD and 20/20 OS. Slit lamp examination was normal and fundus showed a bilateral vitreous haze, predominating on the right eye with papillary edema. Macular OCT revealed a cystoid macular edema of the right eye (Fig. 2A), and a normal macular profile of the left eye (Fig. 2C). Fluorescein angiography revealed a venous non-occlusive and multifocal vasculitis in the right eye (Fig. 2B) bilateral papillary leakage (Fig. 2B, D) with mild peripheral vasculitis on the left eye. On the right eye, macular edema was associated with a diffuse macular leakage (Fig. 2B). Cerebrospinal fluid analysis showed an aseptic meningitis: 9 cells/mm3, glucose and protein levels were normal. PCR for herpes group virus and cultures were negative. Genetic analysis was positive for HLA B51. The patient did not fulfill PEDBD criteria as she only presented ocular and neurological features, diagnosis was retained after a multidisciplinary agreement. The patient was treated with intravenous MP pulses followed by tapering oral prednisone, associated with monthly infliximab perfusions and oral azathioprine. She recovered her full visual acuity after 3 months of treatment with a complete resolution of her macular edema and vasculitis. At last follow-up (57 months after onset), she was still in remission (Fig. 2E–H), while she was receiving infliximab, azathioprine and colchicine.

case #3. A Late Fluorescein angiogram showing venous non-occlusive and multifocal vasculitis. Macular edema was associated with a diffuse macular leakage on the right eye (B). C Left eye: late Fluorescein angiogram showing papillary leakage and D normal macular profile on OCT. E–H Twelve months after treatment initiation. Normalization of both fluorescein angiogram and macular OCT
Case 4
A 16-year-old boy, from Algerian descent, presented with severe general state alteration over the last 6 months, accompanied with aphthosis, headache, apathy and catatonia. He consulted at the emergency department because of red eyes and vision loss that appeared 15 days before. Ophthalmological examination showed a bilateral and severe non-granulomatous panuveitis. BCVA was limited to light perception on the right eye and 20/100 on the left eye. Initial fundus photography showed major bilateral papilledema with venous tortuosity and bilateral vitritis (Fig. 3A, B). Fluorescein angiography confirmed these findings (Fig. 3C, D).

case #4. A, B Initial fundus photography showed major bilateral papilledema with venous tortuosity and bilateral vitritis. C, D Fluorescein angiography confirmed these findings. E–H Fundus photography and fluorescein angiogram performed 3 months after treatment initiation: resolution of the inflammation associated with bilateral optic atrophy
Brain magnetic resonance imaging (MRI) showed a cerebral venous thrombophlebitis at the level of the longitudinal superior sinus and the left lateral sinus. He fulfilled PEDBD criteria with oral aphtosis, ocular, neurological and vascular features.
He was initially treated with intravenous MP pulses, infliximab perfusion, oral azathioprine and colchicine. Enoxaparine and aspirin were added for the thrombosis. One week later, intraocular inflammation and thrombophlebitis had waned, but papillary edema persisted on both eyes, with no improvement of visual acuity. After multidisciplinary discussion, he received intravenous cyclophosphamide pulses. Despite a complete resolution of the intraocular inflammation and the cerebral thrombosis, optic nerves atrophy with complete blindness (logMAR3) developed on both eyes (Fig. 3E–H).
Discussion
In this case series of PBD, ocular manifestations were severe, with comparable phenotypes to what is usually encountered in adult BD patients [1]. All patients had posterior segment involvement, including retinal vasculitis, and required intensive systemic immunosuppressive treatments in association with systemic corticosteroids.
The prevalence of ocular manifestations in both BD and PBD is highly variable. It is reported in 9 to 76% of patients in large PBD series [5, 7, 11,12,13,14,15] and in 18 to 75% of adult BD series [2, 14, 16, 17].
Regarding the ocular phenotypes, the ICBD study reported that retinal vasculitis, posterior uveitis, and anterior uveitis occurred in 23%, 38%, and 40% of adult cases, respectively which seems to be comparable with what is reported in PBD [5, 7, 11, 12]. Papilledema may also be present, as in our cases. The latter can be caused by either inflammation, ischemia, or intracranial hypertension secondary to cerebral venous thrombosis [18,19,20,21]. As in adult BD, various unusual ocular presentations have been reported in PBD such as recurrent neuroretinitis [22] or immune keratitis [23]. In both adult and pediatric BD patients, bilateral panuveitis is likely to be the most frequent presentation, occurring in up to half of the patients [19, 24,25,26]. In both adults and children (as in our series) the macula is frequently involved [19, 24,25,26]. Ocular complications seem comparable in PBD and BD ocular involvement cohorts: cataract, optic atrophy and posterior synechiae are the most frequent [19, 24,25,26], occurring approximatively in a third to half of the patients, followed by rarer complications, such as intraocular pressure elevation, retinal detachment, neovascular glaucoma, phthisis bulbi and band keratopathy (Tables 3 and 4) [19, 24,25,26].
Epidemiology of pediatric BD
Age at onset of symptoms also varies among the different PBD cohorts. Koné-Paut et al. and Nanthapisal et al.reported a mean age at onset of BD of 4.9-year-old (0.1 to 15.7 y.o.) and 7.4 ± 4.2 y.o., respectively [2, 7]. In these two studies, family history was presentin 17% and 24% of patients, respectively, a higher rate than in adult BD cohorts, suggesting a stronger familial aggregation in children [2, 7, 11, 14]. Female/male ratio seems similar in BD and PBD [2, 5, 7, 9, 11,12,13,14,15,16,17, 13, 14, 16, 27]. Geographical location and ethnic origins may be involved in clinical phenotypes seen in PBD cohorts [2]. For instance, the prevalence of ocular involvement reported in Iran and Egypt(66% and 76% respectively) [5, 14], seems higher than what was reported in Italy, Taiwan, China and UK (44%, 27%, 9% and 9% respectively) [11,12,13, 15]. BD is typically considered as a polygenic disorder. Zhou et al. reported 6 families with early onset auto-inflammatory disease caused by heterozygous mutation in the TNFAIP3 gene coding for A20 proteins with clinical manifestations mimicking BD. However, a mutation in A20 protein was identified in only one patient in a large BD genome-wide association study [28].
Diagnostic delay and criteria
As seen in our study, delay of diagnosis may be an important issue in PBD, especially when ocular manifestations reveal the disease, as children may not complain in cases of unilateral and painless ocular involvement. As in other inflammatory diseases, diagnostic delay may impact the severity of clinical presentation. In published literature, diagnostic delays for PBD vary from 2.9 ± 3.6 years [12], to 6.0 ± 3.5 years [7]. However, this long diagnostic delay is also found in adults [16].
In our study, three patients met the PEDBD criteria, while one patient was diagnosed on the combination of typical neurological features (cerebellar peduncle) and retinal vasculitis, both of which are highly suggestive of Behçet's disease [29]. However, three patients met the ICBD criteria designed for adult BD [2]. As in our series, in previously published PBD cohorts, ICBD criteria are constantly more sensitive than PEDBD criteria (71 to 97% versus 36 to 69%, respectively) [5, 12, 13, 15]. ICBD and PEDBD criteria comprise 7 and 6 items, respectively, that are weighted in ICBD classification while they’re not in PEDBD criteria [2, 7]. Thus, the presence of uveitis and oral aphtosis is sufficient to diagnose PBD with ICBD but not with PEDBD criteria. ICBD criteria have been developed from larger cohorts comprising both adult and children originating from various countries, with the underlying hypothesis that adult and pediatric disease are similar [2]. While in most of the cases, oral or genital ulcerations are the first symptoms of the disease, scarce information is available about the referring symptom in neither PBD studies nor BD studies [2, 5, 7, 11,12,13,14,15,16,17, 19, 24,25,26].
In cases of isolated ophthalmic or atypical presentations, other cause of posterior uveitis may be considered in the pediatric context, including infectious diseases, such as toxoplasmosis, toxocarosis, and herpesviridae infections, that can be associated with vitritis and retinal necrosis [30]; inflammatory diseases such as (but not limited to) sarcoidosis or Blau Syndrome that can cause posterior uveitis with retinal vasculitis [31]. Uveitis associated with tubulointerstitial nephritis and uveitis syndrome and juvenile idiopathic arthritis are usually limited to the anterior segment [32, 33].
Treatments
Contrarily to adult BD, there is no recommendation available to treat ocular manifestation in PBD. As in our series, other teams used European alliance of associations for rheumatology (EULAR) recommendation derived treatment protocols [11,12,13,14, 34]. Intravenous high dose corticosteroids are helpful in the acute phase, but long-term oral steroids are used with caution in children essentially because of their impact on children’s growth [35]. Immunosuppressive therapies are recommended in combination with steroids in posterior uveitis [34], but data on their efficacy in PBD are scarce. Some small case series reported the efficacy of treatments such as methotrexate, cyclosporine, chlorambucil, azathioprine, sulfasalazine, cyclophosphamide or thalidomide in this context [11,12,13, 25, 36]. Regarding biotherapies, anti-TNF α, mainly infliximab, which is recommended in EULAR guidelines for severe cases has been reported in PBD series [11, 12, 30, 37]. Serious side effects with long-term anti-TNF α use in non-infectious pediatric uveitis remains rare, infliximab antibodies is the most frequent issue [30, 38].
Interferon have been reported in PBD case reports or small case series in association with conventional immunosuppressive treatments for severe and relapsing forms, but adverse effects are frequent (flu-like syndrome especially) and may be severe (lymphopenia, neutropenia, depression) [36, 30, 39, 40]. In our experience, in addition to the choice of the drug itself, the most important elements in managing PBD and other severe inflammatory ocular conditions in children are i) a close collaboration between pediatricians and ophthalmologists to manage systemic manifestations, optimize systemic treatment while monitoring side effects and adherence, and ii) to provide educational support to patients and their family, in order for them to be involved in the therapeutic project [41].
Prognosis
As in adults, visual prognosis of ocular involvement in PBD is guarded. In a cohort of 36 PBD patients with 66 eyes affected by uveitis, and a mean follow up of 84.7 ± 91.5, Tugal-Tuktun et al. reported 23% of eyes with a severe and irreversible visual loss (BCVA below 20/200), and 17% of patients with legal blindness (BCVA below 20/200 on the best eye) [25]. Koné-Paut et al. reported a BCVA < 20/200 in 19% of PBD patients with uveitis in at least one eye, and 3% of patients with legal blindness [42]. In recent adult BD cohorts with uveitis, the proportion of patient with at least one eye with BCVA < 20/200 varies between 28 and 30% [43, 44]. In PBD, the most frequent cause of legal blindness.
Conclusions
To summarize, as in adult, ocular involvement is a frequent and potentially blinding manifestation of BD in children. According to our case series and the literature, pediatric ocular phenotypes seem comparable to those observed in adults. On the other hand, it seems important not to omit the search for a family history that may raise suspicion of an autoinflammatory disease in this specific context. A multidisciplinary evaluation with an early ophthalmological evaluation is absolutely required for any suspicion of PBD.
Availability of data and materials
The datasets supporting the conclusions of this article are included in the tables and the figures.
Abbreviations
- PBD:
-
Pediatric Behçet disease
- BD:
-
Behçet disease
- PEDBD:
-
Pediatric Behçet’s Disease Study
- ICBD:
-
International Criteria for Behçet’s Disease Study
- BCVA:
-
Best-corrected visual acuity
- SUN:
-
Standardization of Uveitis Nomenclature
- OCT:
-
Optical Coherence Tomography
- IRB:
-
Institutional Review Board
- MEFV:
-
Familial Mediterranean Fever
- PFAPA:
-
Periodic fever, aphthous stomatitis, pharyngitis, adenitis
- MRI:
-
Magnetic resonance imaging
- PCR:
-
Polymerase Chain Reaction
- MP:
-
Methylprednisolone
- EULAR:
-
European alliance of associations for rheumatology
References
Yazici Y, Hatemi G, Bodaghi B, et al. Behçet syndrome. Nat Rev Dis Primers. 2021;7(1):67.
Davatchi F, Chams-Davatchi C, Shams H, et al. Behcet’s disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol. 2017;13(1):57–65.
Zouboulis CC, Kötter I, Djawari D, et al. Epidemiological features of Adamantiades-Behçet’s disease in Germany and in Europe. Yonsei Med J. 1997;38(6):411–22.
Karincaoglu Y, Borlu M, Toker SC, et al. Demographic and clinical properties of juvenile-onset Behçet’s disease: a controlled multicenter study. J Am Acad Dermatol. 2008;58(4):579–84.
Shahram F, Nadji A, Akhlaghi M, et al. Paediatric Behçet’s disease in Iran: report of 204 cases. Clin Exp Rheumatol. 2018;36(6 Suppl 115):135–40.
Yildiz, et al. Pediatric Behçet’s Disease. Front Medi. 2021;8:627192.
Koné-Paut I, Shahram F, Darce-Bello M, et al. Consensus classification criteria for paediatric Behçet’s disease from a prospective observational cohort: PEDBD. Ann Rheum Dis. 2016;75(6):958–64.
Koné-Paut I. Behçet’s disease in children, an overview. Pediatr Rheumatol Online J. 2016;14(1):10.
International Team for the Revision of the International Criteria for Behçet’s Disease (ITR-ICBD). The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–47.
Jabs DA, Nussenblatt RB, Rosenbaum JT. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509–16.
Nanthapisal S, Klein NJ, Ambrose N, et al. Paediatric Behçet’s disease: a UK tertiary centre experience. Clin Rheumatol. 2016;35(10):2509–16.
Gallizzi R, Pidone C, Cantarini L, et al. A national cohort study on pediatric Behçet’s disease: cross-sectional data from an Italian registry. Pediatr Rheumatol Online J. 2017;15(1):84.
Hu YC, Yang YH, Lin YT, et al. Clinical manifestations and anti-TNF alpha therapy of juvenile Behçet’s disease in Taiwan. BMC Pediatr. 2019;19(1):232.
Gheita TA, El-Latif EA, El G II, et al. Behçet’s disease in Egypt: a multicenter nationwide study on 1526 adult patients and review of the literature. Clin Rheumatol. 2019;38(9):2565–75.
Zou, et al. Distinct clinical characteristics of pediatric Behçet’s syndrome: A study from a referral center in China. Modern Rhumatol. 2021;31(6):1158–63.
Alpsoy E, Donmez L, Onder M, et al. Clinical features and natural course of Behçet’s disease in 661 cases: a multicentre study. Br J Dermatol. 2007;157(5):901–6.
Zou, et al. Cluster analysis of phenotypes of patients with Behçet’s syndrome: a large cohort study from a referral center in China. Arthritis Res Ther. 2021;23(1):45.
Hacihamdioglu DO, Demiriz M, Sobaci G, et al. Cerebral vein thrombosis in a four year old with Behçet’s disease. Reumatol Clin. 2014;10(4):254–6.
Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, et al. Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol. 2004;138(3):373–80.
Kim S, Kang S, Roh YJ. A case of anterior ischemic optic neuropathy associated with Behcet’s disease. Eye (Lond). 2011;25(3):395–6.
Yamauchi Y, Cruz JM, Kaplan HJ, et al. Suspected simultaneous bilateral anterior ischemic optic neuropathy in a patient with Behçet’s disease. Ocul Immunol Inflamm. 2005;13(4):317–25.
Rabina G, Amarilyo G, Zur D, et al. Recurrent Neuroretinitis: A Unique Presentation of Behçet’s Disease in a Child. Case Rep Ophthalmol. 2020;11(3):516–22.
Jinagal J, Agarwal A, Negi A, et al. Immune keratitis: An unusual primary presentation of neuro-Behçet’s disease. Eur J Ophthalmol. 2019;29(4):Np5–np8.
Citirik, et al. Ocular findings in childhood-onset Behçet disease. J AAPOS. 2009;13(4):391–5.
Tugal-Tutkun I, Urgancioglu M. Childhood-onset uveitis in Behçet disease:a descriptive study of 36 cases. Am J Ophthalmol. 2003;136(6):1114–9.
Yang P, Fang W, Meng Q, et al. Clinical features of chinese patients with Behçet’s disease. Ophthalmology. 2008;115(2):312–8.e4.
Atmaca L, Boyvat A, Yalçındağ FN, et al. Behçet disease in children. Ocul Immunol Inflamm. 2011;19(2):103–7.
Zhou Q, Wang H, Schwartz DM, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2016;48(1):67–73.
Borhani Haghighi A, Pourmand R, Nikseresht AR. Neuro-Behçet disease. A review. Neurologist. 2005;11(2):80–9.
Sardar E, Dusser P, Rousseau A, et al. Retrospective study evaluating treatment decisions and outcomes of childhood uveitis not associated with Juvenile idiopathic arthritis. J Pediatr. 2017;186:131–7.e1.
Amin SR, Pulido JS. Retinal vasculitis, aneurysms, and neovascularization in Blau syndrome. JAMA Ophthalmol. 2013;131(5):677–80.
Amaro D, Carreño E, Steeples LR, Oliveira-Ramos F, Marques-Neves C, Leal I. Tubulointerstitial nephritis and uveitis (TINU) syndrome: a review. Br J Ophthalmol. 2020;104(6):742–7.
Petty RE, Zheng Q. Uveitis in juvenile idiopathic arthritis. World J Pediatr. 2020;16(6):562–5.
Hatemi G, Christensen R, Bang D, et al. 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis. 2018;77(6):808–18.
Aljebab F, Choonara I, Conroy S. Systematic review of the toxicity of long-course oral corticosteroids in children. PLoS One. 2017;12(1):e0170259.
Sharma SM, Dick AD, Ramanan AV. Non-infectious pediatric uveitis: an update on immunomodulatory management. Paediatr Drugs. 2009;11(4):229–41.
Diwo E, Sève P, Trad S, et al. Therapeutic strategy for the treatment of non-infectious uveitis proposed by an expert panel. Rev Med Interne. 2018;39(9):687–98.
Ardoin SP, Kredich D, Rabinovich E, et al. Infliximab to treat chronic noninfectious uveitis in children: retrospective case series with long-term follow-up. Am J Ophthalmol. 2007;144(6):844–9.
Evereklioglu C, Borlu M. Sustained remission after infliximab in a child with vasculitis refractory to conventional immunosuppressives including interferon-alpha. Br J Ophthalmol. 2008;92(8):1034. 148-9.
Pivetti-Pezzi P, Accorinti M, Pirraglia MP, et al. Interferon alpha for ocular Behçet’s disease. Acta Ophthalmol Scand. 1997;75(6):720–2.
Shivpuri A, Turtsevich I, Solebo AL, Compeyrot-Lacassagne S. Pediatric uveitis: Role of the pediatrician. Front Pediatr. 2022;10:874711.
Koné-Paut I, Darce-Bello M, Shahram F, et al. Registries in rheumatological and musculoskeletal conditions. Paediatric Behçet’s disease: an international cohort study of 110 patients One-year follow-up data. Rheumatology (Oxford). 2011;50(1):184–8.
Amer R, Alsughayyar W, Almeida D. Pattern and causes of visual loss in Behçet’s uveitis: short-term and long-term outcomes. Graefes Arch Clin Exp Ophthalmol. 2017;255(7):1423–32.
Pathanapitoon K, Kunavisarut P, Saravuttikul FA, et al. Ocular Manifestations and Visual Outcomes of Behçet’s Uveitis in a Thai population. Ocul Immunol Inflamm. 2019;27(1):2–6.
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
All authors participated in the acquisition of data, designing the analyses, interpreting results and writing the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors confirm that the study and data collection were compliant with the principles of the Declaration of Helsinki; informed consent was obtained from the parents for all patients. The study was performed according to regulations of the local ethics committee. Institutional Review Board (IRB)/Ethics Committee approval was obtained by the French Society of Ophthalmology (IRB 00008855 Société Française d’Ophtalmologie IRB#1).
Consent for publication
The authors confirm that informed consent was obtained from the parents for all patients to publish information or images that could lead to their identification in an online open-access publication.
Competing interests
Casem Azri, Perrine Dusser-Benisty, Laura Eid, Emmanuel Barreau, Isabelle Koné-Paut, Charlotte Borocco, Caroline Galeotti and, Sami Saad have nothing to disclose.
Marc Labetoulle has been an occasional consultant on subjects outside the scope of this work for Alcon, Allergan, Bausch and Lomb, Dompe, Horus, MSD, Novartis, Santen, Shire, and Thea.
Antoine Rousseau has been an occasional consultant on subjects outside the scope of this work for Novartis, Allergan, Pfizer, Shire and Thea.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Azri, C., Dusser, P., Eid, L. et al. Ocular involvement in pediatric Behçet’s disease: is it different than in adults? (a short case series and mini review). BMC Ophthalmol 23, 474 (2023). https://doi.org/10.1186/s12886-023-03197-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-023-03197-5