
Abstract
Background
To identify the disease-causing gene in a Chinese family affected with congenital aniridia.
Methods
Patients underwent systematic ophthalmic examinations such as anterior segment photography, fundus photography, optical coherence tomography, and fundus fluorescein angiography. The proband was screened for pathogenic variants by whole exome sequencing (WES) and copy number variant (CNV) analysis. Real-time quantitative PCR (RT-qPCR) was applied to confirm the CNV results. Breakpoints were identified by long-range PCR followed by Sanger sequencing.
Results
All seven members of this Chinese family, including four patients and three normal individuals, were recruited for this study. All patients showed bilateral congenital aniridia with nystagmus, except the son of the proband, who presented with bilateral partial coloboma of the iris. A novel heterozygous deletion (chr11:31,139,019–31,655,997) containing the 3’ regulatory enhancers of the PAX6 gene was detected in this family. We also reviewed the reported microdeletions downstream of PAX6 in patients with aniridia.
Conclusions
We identified a novel microdeletion, 517 kb in size located about 133 kb downstream of the PAX6 gene, responsible for congenital aniridia in this Chinese family, which expands the spectrum of aniridia-associated mutations in PAX6.
Similar content being viewed by others
Background
Aniridia (OMIM, 106210) is a rare, bilateral, congenital panocular disorder characterized by the partial or complete absence of the iris, with an incidence of approximately one in 40,000–100,000 live births worldwide. About two-thirds of aniridia cases are familial with autosomal dominant inheritance, while the remaining cases are sporadic [1]. In addition to iris hypoplasia appearing in early infancy, congenital aniridia is usually associated with aniridia-associated keratopathy, glaucoma and cataract, resulting in severely reduced vision in the patient’s second or third decade [2]. Aniridia can also occur as part of other syndromes such as WAGR (OMIM, 194072) [3, 4], WAGRO (OMIM, 612469) [5], Peters anomaly (OMIM, 604229) [6, 7], and Gillespie syndrome (OMIM, 206700) [8, 9].
Paired box gene-6 (encoded by PAX6, OMIM, 607108) is a highly conserved transcriptional regulator that plays a key role in normal ocular and neural development [6]. It is 22 kb in size and locates on chromosome 11p13, but has regulatory regions spanning ~ 450 kb of genomic DNA [10]. Heterozygous mutations in the PAX6 gene or its regulatory regions cause aniridia [11, 12]. To date, more than 600 variants scattered throughout PAX6 locus have been identified, according to the PAX6 gene database (LOVD, https://www.lovd.nl/). Of note, chromosomal rearrangements in the 11p13 region are frequently identified in aniridia cases, and these are thought to affect 3′ regulatory enhancers of the PAX6 gene and influence the expression level of the PAX6 protein [13].
In this study, we identified a novel microdeletion with a length of 517 kb downstream of the PAX6 gene in a Chinese family with congenital aniridia, although two copies of PAX6 are intact. This 11p13 microdeletion contained essential conserved enhancer elements of PAX6, which was likely to be the cause of the familial aniridia in this family.
Methods
Subject recruitment and clinical assessment
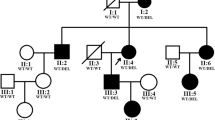
This study was conducted in accordance with the Declaration of Helsinki and with the approval of the medical ethics committee of Shanghai General Hospital, China (Approval No.2020SQ328). A three-generation family with aniridia was recruited and seven family members of this family (Fig. 1) participated in this study. All research subjects provided informed consent. Three of the seven family members were diagnosed with congenital aniridia, and one was with iris coloboma. There was no consanguinity present in this family. Complete and comprehensive clinical and ophthalmic examinations were conducted for each participant, including best corrected visual acuity (BCVA) by Snellen visual acuity, non-contact tonometer, non-contact intraocular pressure (IOP) measurement (TX-20, Canon), slit lamp biomicroscopy, anterior segment photography, fundus photography (200TX, OPTOS), optical coherence tomography (Spectralis, Heidelberg Engineering), fundus fluorescein angiography (Optomap plus, OPTOS), and indocyanine green angiography.

Pedigree of the three-generation family with congenital aniridia. Squares and circles indicate males and females respectively. Solid symbols indicate affected individuals, and open symbols indicate unaffected individuals. The arrow indicates the proband of this family
Whole-exome sequencing (WES)
The WES of the gDNA samples was performed as previously described [14, 15]. The genomic DNA was extracted from peripheral blood samples of the members (Qiagen GmbH, Hilden, Germany). The DNA fragments were hybridized and captured by IDT’s xGen Exome Research Panel V1.0 (Integrated DNA Technologies, San Diego, USA) according to the manufacturer’s Protocol. Novaseq.6000 platform (Illumina, San Diego, USA), with 150 bp pair-end sequencing mode, was used for sequencing the genomic DNA of the proband. Raw image files were processed using CASAVA v1.82 for base calling and generating raw data. The raw reads were aligned to the human reference genome (GRCh38/hg38) using the Burrows Wheeler Aligner tool. Duplicates of the reads were marked and removed using Picard (http://broadinstitute.github.io/picard).
Single-nucleotide variants (SNVs) and InDels (Insertions and Deletions) were called by GATK4-HaplotypeCaller. The frequencies of all SNVs and InDels are annotated using Genome Aggregation Database (gnomAD), the 1000 Genome project (http://brower.1000genomes.org), and the China Metabolic Analytics Project (ChinaMAP) to filter the common variants, with an allele frequency cutoff of 5%. Pathogenicity of remaining variants is predicted by proper analytic tools, including PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/).
For CNV analysis, we applied eXome-Hidden Markov Model (XHMM, zzz.bwh.harvard.edu/xhmm/) principal component analysis (PCA) to normalize the read depth and remove the sequencing noise, and used CNVKit (www.github.com/etal/cnvkit) to perform GC and bias correction. CNV calculation and identification were performed by XHMM Hidden Markov Model (HMM). Variant and CNV filtering was performed as illustrated in Supplementary Figure 1. Detailed information of WES coverage and sequencing depth was listed in Supplementary Table 1.
Copy number variant (CNV) validation by real-time quantitative PCR and breakpoints identification by long-range PCR
CNVs were further validated by real-time quantitative PCR (RT-qPCR). RT-qPCR was accomplished using the SYBR Premix Ex Taq (Takara, Japan) in the ABI PRISM® 7300 real-time-PCR system (Applied Biosystem, Foster City, CA, USA). GAPDH was used as an endogenous control. The relative copy number was computed using 2−ΔΔCt method. Then to define the breakpoint, fragments surrounding the breakpoints were amplified by long-range PCR and Sanger sequencing (Huagene, Shanghai, China). The sequences of the primers are listed in Supplementary Table 2. To define the genomic architecture in triggering genomic rearrangement events, upstream and downstream regions around the breakpoint were analyzed using the RepeatMasker track (http://www.repeatmasker.org/) of UCSC Genome Browser (https://genome.ucsc.edu/).
Results
Clinical findings
In this study, we identified a three-generation family with congenital aniridia including four affected members, as shown in Fig. 1. All three adult-affected patients (I:2, II:1, and II:2) presented with low visual acuity, ranging from 0.2 to 0.3. The Intraocular pressure of the three adult patients was normal (I:2, II:1, and II:2). These three adult patients (I:2, II:1, and II:2) showed phenotypes of bilateral aniridia and nystagmus, while one patient (III:1) was found to suffer from bilateral partial coloboma of the iris (Table 1). Representative photos from anterior segment photography of II:2 (proband) and III:1 are shown in Fig. 2. Mild vitreous opacity, cystoid macular edema, and leakage were observed in the right eye of the proband (Fig. 2). No other ophthalmic phenotypes, such as foveal hypoplasia, microphthalmia, lens defects, and non-ocular abnormalities, such as mental retardation, emotional disorders, renal diseases, and neurological deficits were detected in the four patients. No other family members had aniridia phenotypes or other relevant eye diseases.

Representative photos of ophthalmological findings. A Anterior segment photograph of the proband (II:2) performed aniridia with peripheral iris remnants in both eyes. B Anterior segment photograph of the patient (III:1) displayed bilateral partial coloboma of iris. C, F No obvious abnormalities were found in the fundus images of the proband except mild vitreous opacity in the right eye. D, G Optical coherence tomography (OCT) of the proband displayed cystoid macular edema in the right eye. E, H Fundus angiography of the proband showed slight leakage in the right macular and the optic disc. OD, oculus dexter; OS, oculus sinister
Genetic analysis
To determine the disease-causing gene in this family, whole exome sequencing (WES) was performed on genomic DNA from the peripheral blood of the proband. No possible pathogenic single nucleotide variants (SNVs) and indels related to aniridia were detected in the proband, according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) [16]. However, a possible pathogenic copy number variant (CNV) was detected by XHMM V1.0. A heterozygous deletion of approximately 410 kb (chr11:31,241,412–31,650,221) was found, which covered adjacent genes at the 3’ end of PAX6 including exons 1–9 of doublecortin domain containing 1 (DCDC1), DnaJ heat shock protein family (Hsp40) member C24 (DNAJC24), inner mitochondrial membrane peptidase subunit 1 (IMMP1L), and exons 1–9 of elongator acetyltransferase complex subunit 4 (ELP4).
Identification of a CNV in this family
To confirm the CNV detected by WES in this family, we performed real-time quantitative PCR (RT-qPCR) to examine the copy number of affected exons. The relative copy number of the exons 1–9 of DCDC1, DNAJC24, IMMP1L, and exons 1–9 of ELP4 in the proband was half that in a healthy control, which is consistent with the results of WES (Fig. 3).

CNV confirmation by RT-qPCR in the proband of this Family. The relative copy number of exons 1 − 20 of DCDC1, DNAJC24, IMMP1L, ELP4, and PAX6 in the proband of this Family. The affected exons and genes were colored in red
The sizes of intron 9 of DCDC1 and intron 9 of ELP4 are both longer than 100 kb, which are too large to amplify by long-range PCR. To further define the breakpoints of the CNV, we used RT-qPCR to identify the deleted regions in intron 9 of DCDC1 and intron 9 of ELP4 (Fig. 4A and B). Then, we performed long-range PCR with primers F1 and R10. An approximately 3,000 bp fragment was amplified in the genomic DNA from the proband (II:2) and her son (III:1), but not from a healthy control (Fig. 4C). Original gels are presented in Supplementary Figure 2. Sanger sequencing of the PCR products identified a 516,979 bp deletion (chr11:31,139,019–31,655,997) between the proximal and distal breakpoints in II:2 and III:1. The proximal breakpoint of this microdeletion was approximately 123 kb from the 3’ end of the PAX6 and was located in the HS234 fragment (hypersensitive sites 2, 3, and 4) (chr11: 31,652,512–31,657,084) in intron 9 of ELP4. This microdeletion involved three conserved regulatory elements (RB, E180B, and HS) of PAX6 and two H3K27ac enrichment sites (Fig. 4D).

Identification of the breakpoints of CNV in this family. A The relative copy number of different regions in intron 9 of DCDC1. B The relative copy number of different regions in intron 9 of ELP4. C Long-range PCR by primer pair F1/R10 on gDNA from the proband. An approximate 3,000 bp fragment was amplified in II:2 and III:1, but not in a healthy control. D Sequence chromatograms showing the microdeletion (chr11:31,139,019–31655997) (hg38) identified in this study
To investigate whether repetitive elements intersected with the breakpoints, which may lead to genomic instability and CNVs [17], we used RepeatMasker to align the breakpoints with specific repetitive elements. Unfortunately, the breakpoints were located in regions without repeated elements, indicating that other mechanisms may be involved in the genomic arrangement in this family. Taken together, a 517 kb heterozygous deletion containing four annotated genes, DCDC1, DNAJC24, IMMP1L, and ELP4, was identified in this Chinese family with congenital aniridia, suggesting that transcription-regulating elements in the deleted region are required for the expression of PAX6.
Overview of previously reported microdeletions excluding the PAX6 gene
Microdeletions involving the 11p3q region are frequent in cases with congenital aniridia, and these microdeletions partly or completely encompass the PAX6 gene or remove the PAX6 gene. [2, 4, 13]. To date, 30 genomic rearrangements downstream of the PAX6 have been reported in the literature to cause aniridia without neurodevelopment disorders (Fig. 5 and Supplementary Table 3) [18,19,20,21,22,23,24,25,26,27,28,29,30,31]. These deleted regions range from 49 to 1,300 kb, and the distance from the 3’ of PAX6 range from 1 to 467 kb. Among them, 29 microdeletions entirely or partially spanned the critical region, which is required for the PAX6 gene transcription, as suggested by Ansari et al. 2016 [9].

Schematic diagram of 11p13 microdeletions involving regulatory regions 3’ of PAX6 identified in patients with aniridia. Genes are represented by grey and black boxes and arrows indicate the direction of transcription. Known PAX6 enhancers, RB, E180B, HS, SIMO, E120, E100, and E60 are indicated by green oval shapes. Horizontal blue bars represent microdeletions in 11p13 identified in aniridia patients with the integrity of PAX6. The vertical dashed lines indicated the “critical region” required for the Pax6 gene transcription, which was defined first as 244 kb long by Ansari et al. Genomic coordinates are based on human genome assembly hg38
Discussion
Congenital aniridia is a severe bilateral panocular visual disorder with an autosomal dominant inheritance. PAX6, a highly conserved transcription factor, plays a fundamental role in organogenesis by regulating downstream target genes [6, 32, 33]. Many studies [34,35,36,37,38,39] have reported that mutations in the PAX6 gene and its regulatory regions are the leading cause of congenital aniridia. In this study, the novel 517 kb heterozygous deletion (chr11:31,139,019–31,655,997) downstream of PAX6 containing four annotated genes, DCDC1, DNAJC24, IMMP1L, and ELP4, is likely to be the cause of the familial aniridia in this Chinese family. This finding further demonstrates the importance of 3’ downstream regulatory elements on the expression of PAX6 and broadens the spectrum of PAX6 mutations resulting in congenital aniridia.
Defects in PAX6 lead to a broad range of clinical phenotypes, with the most common being aniridia. In addition to varying degrees of iris defects, other associated ocular phenotypes (e.g. microphthalmia, optic nerve anomalies, and anterior segment dysgenesis) [40, 41] and systemic abnormalities (e.g. hormonal, metabolic, gastrointestinal, genitourinary, and neurologic disorders) [3,4,5,6,7,8,9] have been reported in some cases with PAX6 mutations. Moreover, disruption of nearby genes (ELP4, IMMP1L, DCDC1, and DNAJC24) may contribute to a range of neurodevelopmental phenotypes [19, 26, 42, 43]. However, in this three-generation family, no other obvious ocular or non-ocular abnormalities were observed in the four patients with congenital aniridia, which is consistent with previous studies showing that defects in the 3’-cis-regulatory regions are generally associated with a milder phenotype without keratopathy, nystagmus, or foveal hypoplasia [26, 30]. Of note, attention should be paid to mild iridopathy in III:1, which may lead to potential blindness and more serious phenotypes in later generations.
PAX6 is known to be affected by a downstream cluster of ultraconserved transcriptional regulatory elements, such as RB, E180B, HS, SIMO, E100, and E60, which play roles as important enhancers in regulating normal PAX6 expression by transgenic reporter studies [29, 44]. RB, the most distal PAX6 enhancer, contributes to its expression in prosomere P2 and the pineal gland [45]. E180B drives the expression in the trigeminal nerves and spinal nerve tracts [46]. DNase hypersensitivity cluster (HS1-8), a marker for active cis-regulatory sequences, is located 130 kb downstream of the PAX6 poly(A) site [47, 48]. Deletion of HS6 results in a complete loss of expression in the pre-cerebellar neuroepithelium, pontine migratory streams, and pre-cerebellar nuclei. Separating of HS5 abolishes expression in the lateral band around the thalamus. HS234, a ~ 4 kb fragment, recapitulated distinct parts of the endogenous expression pattern of PAX6 [44]. Therefore, the defects of RB, E180B, and HS elements are likely to be the underlying cause of familial aniridia by affecting the PAX6 expression in this Chinese family. Further studies are needed to verify their transcription-regulating capability of PAX6.
Conclusion
In conclusion, a novel 517 kb heterozygous deletion of chromosome 11p13 downstream of the PAX6 is likely to be the underlying cause of iris abnormalities in this family. These results enrich the mutation spectrum of PAX6, providing further evidence that genetic defects in the 3’ regulatory elements downstream region of PAX6 lead to congenital aniridia.
Availability of data and materials
The datasets generated during the current study are available in the Sequence Read Archive (SRA) repository, the accession number is SRR21619456 and the persistent web link is https://www.ncbi.nlm.nih.gov/sra/?term=SRR21619456.
Abbreviations
- PAX6:
-
Paired box gene-6
- WES:
-
Whole exome sequencing
- CNV:
-
Copy number variant
- RT-qPCR:
-
Real-time quantitative PCR
- WAGR:
-
Wilms tumor, aniridia, genitourinary anomalies, and mental retardation syndrome
- WAGRO:
-
WAGR Syndrome with obesity
- OCT:
-
Optical coherence tomography
- BCVA:
-
Best corrected visual acuity
- IOP:
-
Intraocular pressure
- NA:
-
Not available
- SNV:
-
Single nucleotide variant
- ACMG:
-
American College of Medical Genetics and Genomics
- DCDC1:
-
Doublecortin domain containing 1
- DNAJC24:
-
DnaJ heat shock protein family (Hsp40) member C24
- IMMP1L:
-
Inner mitochondrial membrane peptidase subunit 1
- ELP4:
-
Elongator acetyltransferase complex subunit 4
- HS:
-
Hypersensitive sites
References
Hu DN. Prevalence and mode of inheritance of major genetic eye diseases in China. J Med Genet. 1987;24(10):584–8.
Wawrocka A, Krawczynski MR. The genetics of aniridia - simple things become complicated. J Appl Genet. 2018;59(2):151–9.
Jung R, Rauch A, Salomons GS, Verhoeven NM, Jakobs C, Michael Gibson K, Lachmann E, Sass JO, Trautmann U, Zweier C, et al. Clinical, cytogenetic and molecular characterization of a patient with combined succinic semialdehyde dehydrogenase deficiency and incomplete WAGR syndrome with obesity. Mol Genet Metab. 2006;88(3):256–60.
Lim HT, Kim DH, Kim H. PAX6 aniridia syndrome: clinics, genetics, and therapeutics. Curr Opin Ophthalmol. 2017;28(5):436–47.
Brémond-Gignac D, Crolla JA, Copin H, Guichet A, Bonneau D, Taine L, Lacombe D, Baumann C, Benzacken B, Verloes A. Combination of WAGR and Potocki-Shaffer contiguous deletion syndromes in a patient with an 11p11.2-p14 deletion. Eur J Hum Genet. 2005;13(4):409–413.
Kokotas H, Petersen MB. Clinical and molecular aspects of aniridia. Clin Genet. 2010;77(5):409–20.
François J, Lentini F, de Rouck F. Gillespie’s syndrome (incomplete aniridia, cerebellar ataxia and oligophrenia). Ophthalmic Paediatr Genet. 1984;4(1):29–32.
Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics. 2005;116(4):984–8.
Ansari M, Rainger J, Hanson IM, Williamson KA, Sharkey F, Harewood L, Sandilands A, Clayton-Smith J, Dollfus H, Bitoun P, et al. Genetic Analysis of “PAX6-Negative” Individuals with Aniridia or Gillespie Syndrome. PLoS ONE. 2016;11(4): e0153757.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012;20(10):1011–7.
Ton CC, Hirvonen H, Miwa H, Weil MM, Monaghan P, Jordan T, van Heyningen V, Hastie ND, Meijers-Heijboer H, Drechsler M, et al. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell. 1991;67(6):1059–74.
Glaser T, Walton DS, Maas RL. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992;2(3):232–9.
Landsend ES, Utheim ØA, Pedersen HR, Lagali N, Baraas RC, Utheim TP. The genetics of congenital aniridia-a guide for the ophthalmologist. Surv Ophthalmol. 2018;63(1):105–13.
Liang J, She X, Chen J, Zhai Y, Liu Y, Zheng K, Gong Y, Zhu H, Luo X, Sun X. Identification of novel PROM1 mutations responsible for autosomal recessive maculopathy with rod-cone dystrophy. Graefes Arch Clin Exp Ophthalmol. 2019;257(3):619–628.
Wang Y, Liu S, Zhai Y, Liu Y, Wan X, Wang W, Wang F, Sun X. Identification of a novel RPGR mutation associated with X-linked cone-rod dystrophy in a Chinese family. BMC Ophthalmol. 2021;21(1):401.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Garg P, Martin-Trujillo A, Rodriguez OL, Gies SJ, Hadelia E, Jadhav B, Jain M, Paten B, Sharp AJ. Pervasive cis effects of variation in copy number of large tandem repeats on local DNA methylation and gene expression. Am J Hum Genet. 2021;108(5):809–24.
Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T. 3’ deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci USA. 2000;97(25):13755–9.
Davis LK, Meyer KJ, Rudd DS, Librant AL, Epping EA, Sheffield VC, Wassink TH. Pax6 3’ deletion results in aniridia, autism and mental retardation. Hum Genet. 2008;123(4):371–8.
Bayrakli F, Guney I, Bayri Y, Ercan-Sencicek AG, Ceyhan D, Cankaya T, Mason C, Bilguvar K, Bayrakli S, Mane SM, et al. A novel heterozygous deletion within the 3’ region of the PAX6 gene causing isolated aniridia in a large family group. J Clin Neurosci. 2009;16(12):1610–4.
Cheng F, Song W, Kang Y, Yu S, Yuan H. A 556 kb deletion in the downstream region of the PAX6 gene causes familial aniridia and other eye anomalies in a Chinese family. Mol Vis. 2011;17:448–55.
Zhang X, Zhang Q, Tong Y, Dai H, Zhao X, Bai F, Xu L, Li Y. Large novel deletions detected in Chinese families with aniridia: correlation between genotype and phenotype. Mol Vis. 2011;17:548–57.
Simioni M, Vieira TP, Sgardioli IC, Freitas EL, Rosenberg C, Maurer-Morelli CV, Lopes-Cendes I, Fett-Conte AC, Gil-da-Silva-Lopes VL. Insertional translocation of 15q25-q26 into 11p13 and duplication at 8p23.1 characterized by high resolution arrays in a boy with congenital malformations and aniridia. Am J Med Genet A 2012;158a(11):2905–2910.
Wawrocka A, Budny B, Debicki S, Jamsheer A, Sowinska A, Krawczynski MR. PAX6 3’ deletion in a family with aniridia. Ophthalmic Genet. 2012;33(1):44–8.
Guo H, Dai L, Huang Y, Liao Q, Bai Y. A large novel deletion downstream of PAX6 gene in a Chinese family with ocular coloboma. PLoS ONE. 2013;8(12): e83073.
Addis L, Ahn JW, Dobson R, Dixit A, Ogilvie CM, Pinto D, Vaags AK, Coon H, Chaste P, Wilson S, et al. Microdeletions of ELP4 Are Associated with Language Impairment, Autism Spectrum Disorder, and Mental Retardation. Hum Mutat. 2015;36(9):842–50.
Blanco-Kelly F, Palomares M, Vallespín E, Villaverde C, Martín-Arenas R, Vélez-Monsalve C, Lorda-Sánchez I, Nevado J, Trujillo-Tiebas MJ, Lapunzina P, et al. Improving molecular diagnosis of aniridia and WAGR syndrome using customized targeted array-based CGH. PLoS ONE. 2017;12(2): e0172363.
Plaisancié J, Tarilonte M, Ramos P, Jeanton-Scaramouche C, Gaston V, Dollfus H, Aguilera D, Kaplan J, Fares-Taie L, Blanco-Kelly F, et al. Implication of non-coding PAX6 mutations in aniridia. Hum Genet. 2018;137(10):831–46.
Syrimis A, Nicolaou N, Alexandrou A, Papaevripidou I, Nicolaou M, Loukianou E, Christophidou-Anastasiadou V, Malas S, Sismani C, Tanteles GA. Aniridia due to a novel microdeletion affecting PAX6 regulatory enhancers: case report and review of the literature. J Genet. 2018;97(2):555–62.
Vasilyeva TA, Marakhonov AV, Voskresenskaya AA, Kadyshev VV, Käsmann-Kellner B, Sukhanova NV, Katargina LA, Kutsev SI, Zinchenko RA. Analysis of genotype-phenotype correlations in PAX6-associated aniridia. J Med Genet. 2021;58(4):270–4.
Wawrocka A, Walczak-Sztulpa J, Socha M, Kuszel L, Sowinska-Seidler A, Budny B, Bukowska-Olech E, Pilas-Pomykalska M, Jamsheer A, Krawczynski MR. Homozygous microdeletion in the 11p13 region in the patient with isolated form of aniridia: New challenges in the genetic diagnostics of aniridia. Am J Med Genet A. 2022;188(2):642–7.
Azuma N, Yamaguchi Y, Handa H, Hayakawa M, Kanai A, Yamada M. Missense mutation in the alternative splice region of the PAX6 gene in eye anomalies. Am J Hum Genet. 1999;65(3):656–63.
Georgala PA, Carr CB, Price DJ. The role of Pax6 in forebrain development. Dev Neurobiol. 2011;71(8):690–709.
Park SH, Kim MS, Chae H, Kim Y, Kim M. Molecular analysis of the PAX6 gene for congenital aniridia in the Korean population: identification of four novel mutations. Mol Vis. 2012;18:488–94.
Cai F, Zhu J, Chen W, Ke T, Wang F, Tu X, Zhang Y, Jin R, Wu X. A novel PAX6 mutation in a large Chinese family with aniridia and congenital cataract. Mol Vis. 2010;16:1141–5.
Chen P, Zang X, Sun D, Wang Y, Wang Y, Zhao X, Zhang M, Xie L. Mutation analysis of paired box 6 gene in inherited aniridia in northern China. Mol Vis. 2013;19:1169–77.
Lee PC, Lam HH, Ghani SA, Subrayan V, Chua KH. Investigation of a PAX6 gene mutation in a Malaysian family with congenital aniridia. Genet Mol Res. 2014;13(2):3553–9.
Liu X, Wu Y, Miao Z, Zhang H, Gong B, Zhu X, Huang L, Shi Y, Hao F, Ma S, et al. A novel deletion downstream of the PAX6 gene identified in a Chinese family with congenital aniridia. Ophthalmic Genet. 2018;39(4):428–36.
Wawrocka A, Sikora A, Kuszel L, Krawczynski MR. 11p13 deletions can be more frequent than the PAX6 gene point mutations in Polish patients with aniridia. J Appl Genet. 2013;54(3):345–51.
Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E, Yamada M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003;72(6):1565–70.
Tzoulaki I, White IM, Hanson IM. PAX6 mutations: genotype-phenotype correlations. BMC Genet. 2005;6:27.
Balay L, Totten E, Okada L, Zell S, Ticho B, Israel J, Kogan J. A familial pericentric inversion of chromosome 11 associated with a microdeletion of 163 kb and microduplication of 288 kb at 11p13 and 11q22.3 without aniridia or eye anomalies. Am J Med Genet A. 2016;170a(1):202–209.
Toral-Lopez J, Huerta LMG, Messina-Baas O, Cuevas-Covarrubias SA. Submicroscopic 11p13 deletion including the elongator acetyltransferase complex subunit 4 gene in a girl with language failure, intellectual disability and congenital malformations: A case report. World J Clin Cases. 2020;8(21):5296–303.
Lima Cunha D, Arno G, Corton M, Moosajee M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes (Basel). 2019;10(12):1050. https://doi.org/10.3390/genes10121050.
Kleinjan DA, Seawright A, Mella S, Carr CB, Tyas DA, Simpson TI, Mason JO, Price DJ, van Heyningen V. Long-range downstream enhancers are essential for Pax6 expression. Dev Biol. 2006;299(2):563–81.
Bhatia S, Monahan J, Ravi V, Gautier P, Murdoch E, Brenner S, van Heyningen V, Venkatesh B, Kleinjan DA. A survey of ancient conserved non-coding elements in the PAX6 locus reveals a landscape of interdigitated cis-regulatory archipelagos. Dev Biol. 2014;387(2):214–28.
Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum Mol Genet. 2001;10(19):2049–59.
McBride DJ, Buckle A, van Heyningen V, Kleinjan DA. DNaseI hypersensitivity and ultraconservation reveal novel, interdependent long-range enhancers at the complex Pax6 cis-regulatory region. PLoS ONE. 2011;6(12): e28616.
Acknowledgements
We thank all family members for their participation, and all the clinicians and medical staff for their assistance in finishing this study.
Funding
This work was supported by the National Natural Science Foundation of China (82000906), Shanghai Sailing Program (19YF1439200), and Science and Technology Innovation Action Plan of Shanghai Science and Technology Commission (2014090067000).
Author information
Authors and Affiliations
Contributions
YL, JC, and YZ collected clinical samples and performed the molecular experiment. TW and ZC collected the blood samples. JC performed interpreted the sequencing data. YL, JC, and YZ wrote the first draft of the article. QS, XW, HL, and XS designed the experiments and corrected the article. All authors reviewed the manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee of Shanghai General Hospital (Approval No.2020SQ328). A written informed consent was obtained from each participant.
Consent for publication
A written informed consent was acquired from each participant for the publication of this research and any related images.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Figure 1. Filtering strategy of variants and CNVs. (A) single nucleotide variants/indels; (B) copy number variants.
Additional file 2:
Supplementary Figure 2. Original gels of Long-range PCR in Fig. 4.
Additional file 3:
Supplementary Table 1. Detailed information of WES coverage and sequencing depth.
Additional file 4:
Supplementary Table 2. Primers used for defining the breakpoint.
Additional file 5:
Supplementary Table 3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Chen, J., Zheng, Y. et al. A novel microdeletion of 517 kb downstream of the PAX6 gene in a Chinese family with congenital aniridia. BMC Ophthalmol 23, 393 (2023). https://doi.org/10.1186/s12886-023-03147-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-023-03147-1