
Abstract
Current immunotherapies are unsatisfactory against uveal melanoma (UM); however, elevated CD8+ T cell infiltration level indicates poor prognosis in UM. Here, we aimed to identify co-expressed gene networks promoting CD8+ T cell infiltration in UM and created a prognostic hazard model based on the identified hub genes. Raw data and clinical information were downloaded from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. Stromal-immune comprehensive score (ESTIMATE) was used to evaluate the immune-infiltration landscape of the tumor microenvironment. Single-Sample Gene Set Enrichment Analysis (ssGSEA) and Weighted Correlation Network Analysis (WGCNA) were used to quantify CD8+ T cell infiltration level and identify hub genes. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed to analyze the biological processes. Least absolute shrinkage and selection operator (LASSO) Cox regression were used to establish a prognostic model, which was further validated. Finally, pan-cancer analysis evaluated these genes to be associated with CD8+ T cell infiltration in other tumors. In conclusion, the proposed four-gene (PTPN12, IDH2, P2RX4, and KDELR2) prognostic hazard model had satisfactory prognostic ability. These hub genes may promote CD8+ T cell infiltration in UM through antigen presentation, and CD8+ T cell possibly function as Treg, resulting in poor prognosis. These findings might facilitate the development of novel immunotherapies.
Similar content being viewed by others

Introduction
Uveal melanoma (UM) is the most common primary intraocular malignancy in adults. It is an aggressive malignancy composed of malignant melanoma cells. More than 90% of UM involves the choroid and usually presents with symptoms such as blurred or distorted vision, visual field loss, or photopsia. UM has a strong tendency to metastasize from the eyes to the liver [1]. Approximately 40% of UM patients have liver metastases, leading to a high mortality rate [2]. Patients receiving UM treatment have a high rate of local tumor control, however, most patients eventually die of metastasis and have a median overall survival (OS) duration of 12 months [3].
The treatment of metastatic UM includes systemic chemotherapy, immunotherapy, and molecular targeted therapy [4]. Among them, immunotherapy has achieved remarkable results in the treatment of various malignant tumors. However, the low mutational burden and low immunogenicity of UM may result in high resistance to immunotherapy, and the response rate to anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) and anti-programmed cell death-1 (PD-1) checkpoint inhibitor therapy is limited [5,6,7]. These immunotherapies cannot prolong the survival period of patients with metastatic UM [8]. Therefore, developing new immunotherapies for metastatic UM has become a priority.
Recently, a new immune-mobilizing monoclonal T-cell receptors (TCR) against cancer (ImmTAC), called Tebentafusp, has recently been confirmed to be the first drug to significantly prolong the survival of patients with metastatic UM [9]. The U.S. Food and Drug Administration (FDA) granted approval to tebentafusp-tebn (Kimmtrak) for the treatment of adult patients with HLA-A*02:01–positive, unresectable or metastatic uveal melanoma. As described in the FDA’s approval of Tebuconazole, this drug that pairs an anti-CD3 immune effector cell-binding domain with a high-affinity gp100-directed TCR is not an option for every patient with advanced uveal melanoma. Because the immune-mobilizing TCR portion of tebentafusp recognizes gp100 peptides presented only on the HLA-A*02:01 molecule, the coverage of this therapy is limited to 40% of individuals worldwide [9, 10]. Furthermore, although tebentafusp extended overall survival at 1 year to 73% and median survival to 21.7 months in metastatic UM patients, the prognosis of metastatic UM remains poor compared to other cancers, therefore, new immunotherapeutic approaches still need to be explored in order to further prolong survival time of patients [11].
As the main component of the tumor microenvironment (TME), immune infiltrates contribute to tumor progression and immunotherapy response [12]. Tumor infiltrating lymphocytes (TILs) are polymorphic, mainly including T, B, NK, and myeloid cells [13]. TILs in uveal melanoma are predominantly CD8+ T cells; the frequency of CD4+ T cells is low, and B cells and NK cells are only rarely identified [14,15,16]. In particular, CD8+ T cells play a central role in cancer immunity by recognizing specific antigenic peptides on the surface of target cells through the TCR, thus killing malignant cells [17]. Therefore, T lymphocyte infiltration in a variety of tumor biopsy samples has been associated with improved survival in a series of retrospective studies of patients with cancer [18,19,20]. However, several studies have shown that higher TIL infiltration and lower PD-L1 expression in UM appear to be associated with poor prognosis [21,22,23]. These findings are contrary to other studies where higher PD-L1 expression was correlated with poor outcome in solid tumors including cutaneous melanoma [24, 25]. It could be one of the reasons for the ineffectiveness of existing immunotherapies against UM. Therefore, identification of tumor-infiltrating CD8+ T cell-related genes may provide ideas for the development of new immunotherapies applicable to UM.
In this study, we used bioinformatic methods to identify co-expressed gene networks promoting CD8+ T cell infiltration in UM. Then, a prognostic hazard model based on the hub genes was constructed. We further analyzed the biological processes involved in these hub genes by GO, KEGG, and GSEA, and proposed new ideas for treating UM based on these genes and biological processes. This study could be helpful to achieve the goal of enhancing the efficacy of immunotherapy against UM.
Materials and methods
Data source
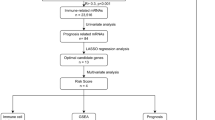
First, RNA sequencing (RNA-seq) and paired clinical data of UM were downloaded from the TCGA database (https://portal.gdc.cancer.gov/). The RNA-seq transcriptome data (in FPKM) were annotated using the human General Transfer Format (human.gtf) from the Ensembl database (https://www.ensembl.org/) with the Strawberry Perl software (version 5.28.2.1). Second, gene expression microarray and clinical data from the GSE44295 and GSE84976 dataset were downloaded as a series of matrix files from the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) database. Probe IDs were matched to gene symbols using the GPL6883 and GPL10558 platform. The mean expression value of the probes was used as the expression value for the gene in question if multiple probes were mapped to a single gene. A detailed work flow chart of this research displaying our entire analysis process is shown in Fig. 1. All methods were performed in accordance with the guidelines and regulations in the guidelines for analysis of RNA sequencing data published by Koch et al. and Chen et al. [26, 27].

The work flow chart of this research
Immune cell infiltration analysis
The 29 immune-associated marker gene sets were obtained from a previous study [28]. The enrichment scores of the 29 immune-associated gene sets in each UM sample were quantified by single-sample gene set enrichment analysis (ssGSEA) using the “GSVA” R package. To identify gene signatures expressed by immune cell populations and immune pathways, ssGSEA was applied to every tumor sample.
Tumor microenvironment (TME)
We obtained the TME of UM cases from the TCGA database. Immune (immune cell infiltration), stromal (stromal content), and stromal-immune comprehensive score (ESTIMATE) scores, as well as tumor purity were calculated for every UM sample using the ESTIMATE algorithm integrated in the “estimate” R package [29]. In addition, TCGA-UM SNP data were obtained, and the tumor mutation burden of each sample was evaluated.
Weighted correlation network analysis (WGCNA)
WGCNA is a data mining method especially designed for studying biological networks based on pairwise correlations between variables. WGCNA was used to identify co-expression gene modules, explore associations between gene networks and CD8+ T cell infiltration levels. After WGCNA analysis, modules that are highly correlated with clinical features are defined as hub modules, and genes with high correlation in hub modules are hub genes [30, 31]. The soft threshold power of β was calculated following the scale-free topological criterion, and a weighted adjacency matrix was generated. The number of genes in the minimum module was set as 100. In addition, modules with a distance of less than 0.25 were merged. CD8+ T cell infiltration levels, stromal score, immune score, tumor purity, and TMB were used as input files for correlation analysis between modules and phenotypes. We identified the hub genes with module membership (MM) > 0.5 and gene significance (GS) > 0.2 in the co-expressed modules having the highest correlation with CD8+ T cell infiltration.
Enrichment analysis and protein-protein interaction (PPI) network of hub genes
We converted the hub gene id to entrez ID and performed GO (http://geneontology.org/) enrichment analysis to explore the biological function, cell composition, and molecular function of the selected hub genes. Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed to explore significant enrichment pathways. The recurring instances of neighboring genes (STRING) online search tool (version 11.5) was used to construct a PPI network (minimum required interaction score > 0.7) [32]. Then, Cytoscape software (version 3.9.0) was used to build the final PPI network [33].
Construction of the gene-related prognostic model
Univariate Cox regression analysis was used to identify genes significantly related to prognosis, and least absolute shrinkage and selection operator (LASSO) COX regression analysis was applied to further narrow the scope of UM marker genes and create a prognostic hazard model by “glmnet” and “survival” R packages. Kaplan–Meier analysis and area under the curves (AUCs) were used to evaluate the accuracy of the prognostic hazard model in the TCGA-UM, GSE44295 and GSE84976 cohorts respectively. Finally, univariate and multivariate Cox regression analyses were used to evaluate whether the constructed prognostic hazard model can be used as an independent prognostic factor.
Gene set enrichment analysis (GSEA)
The gene matrix in TCGA-UM was divided into low- and high-expression groups according to the median expression levels of hub genes, and the GSEA software (version 4.1.0) was used to determine the biological functions of these genes.
Pan-cancer analysis
Tumor Immunity Estimation Resource (TIMER2.0; http://timer.cistrome.org/) was used to analyze the correlation between the derived hub genes in UM and CD8+ T cell infiltration levels in 32 additional cancers [34].
Statistical analysis
All statistical analyses were performed using R version 4.1.1 software (http://www.r-project.org/) and Sangerbox tools (http://www.sangerbox.com/tool). Statistical significance was set at p < 0.05.
Results
Identification of CD8+ T cell infiltration-related modules in UM
To quantify the levels of immune cell infiltration in the TCGA-UM cohort, we used ssGSEA to calculate the enrichment score for all immune cells in each sample. The TCGA cohort was divided into low- and high-immunity groups by applying unsupervised consensus clustering analysis. To validate the immunity of immune subgroups, we show the TME in the heatmap (Fig. 2). The high-immunity group had higher CD8+ T cell enrichment score and lower tumor purity. Then, WGCNA was performed to identify hub genes and co-expression modules with similar expression profiles related to CD8+ T cell infiltration in UM. To build a scale-free network, a value of β = 4 was identified as a soft threshold. We clustered the genes of 80 samples and constructed a hierarchical clustering tree using the dynamic hybrid cutting method, and finally generated 28 co-expression models. Each leaf on the tree represents a gene, and genes with similar expression patterns are grouped together to form a branch of the tree, representing a gene module. Finally, Spearman correlation analysis was used to evaluate the correlation between modules and phenotypes (Fig. 3a). Among them, the pink module containing 240 genes had the highest correlation with CD8+ T cell infiltration (r = 0.54) and was the most significant module (p = 2e-7); thus, it was selected as the hub module. These immune-associate phenotypes were defined as gene significance (GS). The MM and GS correction analyses of the pink module are shown in Fig. 3b. GS and MM were closely correlated in the pink module for UM tissue, reflecting the strong correlation between UM tissue and the pink module genes.

ssGSEA results, immune score, estimated score, stroma score, and tumor purity of samples in TCGA-UM.

The result of WGCNA analysis. a Correlation between different phenotypes and co-expression modules. b Correlation of genes in pink modules with CD8+ T cell levels, immune score, estimated score, stroma score, and tumor purity
Functional analysis of the CD8+ T cell-infiltration related hub Module
To explore the underlying mechanism and biological functions of the CD8+ T cell infiltration-related module, 61 hub genes of the pink module were extracted for GO term and KEGG pathway annotation analyses. GO analysis identified “antigen processing and presentation of peptide antigen” as the most significant biological processes (Fig. 4a). KEGG analysis identified pathways significantly related to immunity, including antigen processing and presentation (Fig. 4b). To explore the interaction between hub genes, a PPI network was constructed using the STRING database and Cytoscape software (Fig. 4c).

The result of GO and KEGG analysis (a) GO enrichment analysis of pink module; (b) KEGG enrichment analysis of pink module. (c) Protein-protein network of the pink module
Construction of a prognostic hazard model
To further identify CD8+ T cell infiltration-related biomarkers that could predict UM prognosis, univariate Cox regression and LASSO Cox regression were used to analyze hub genes in the pink module. First, univariate Cox regression analysis was used to identify genes related to prognosis among the CD8+ T cell infiltration-related hub genes in the pink module (Fig. 5a). Then, Lasso Cox regression analysis based on 19 eligible genes (p < 0.05) was used to construct a prognostic hazard model (Fig. 5b, c). TCGA and GEO data were used as the training and test datasets, respectively. As a result, we obtained four prognostic genes for constructing the model. The risk score was calculated by summing the weighted gene expression level of each of the four genes multiplied by their respective LASSO coefficients: Risk score = 0.00399 * PTPN12 + 0.51624 * IDH2 + 0.47885 * P2RX4 + 0.08205 * KDELR2. The constructed hazard model was used to calculate the risk score of every sample, and the samples were divided into high- and low-risk groups according to the Youden index. The prognosis of the high-risk group was significantly worse than that of the low-risk group (p < 0.001). In addition, AUCs were drawn to assess the prognostic power of the risk characteristics. The AUCs of the prognostic hazard model corresponding to 1-, 3-, and 5-year overall survival (OS) were 0.808, 0.974, and 1.000, respectively, which demonstrates the high accuracy of this model (Fig. 5d). As shown in Fig. 5e,f, the OS of the low-risk group from the GEO dataset was significantly higher than that of the high-risk group (p = 0.023, p = 0.005). This result is consistent with our previous findings in the TCGA cohort. This four-gene prognostic hazard model corresponds to AUCs of 0.602, 0.655 and 0.564 for 1-year, 3-year and 5-year OS in GSE44295 and 0.842 and 0.834 for 3-year and 5-year OS in GSE84976, respectively, which further confirms that this model has high sensitivity and specificity and can be used as a reliable predictor of OS in patients with UM. Finally, univariate Cox regression analysis showed that age, stage, and risk score had prognostic value, but gender did not. Then, multivariate Cox regression analysis showed that only the risk score can be used as an independent prognostic factor (Fig. 6). Therefore, the risk score can be used as a prognostic factor of patient OS independent of other clinical characteristics.

Construction and validation of prognostic hazard model. a Hazard ratio (HR), 95% confidence interval (CI) of prognostic-related genes calculated by univariate Cox regression. b, c Prognostic hazard model constructed by Lasso regression analysis. d–f Survival analysis and AUCs of TCGA and GEO data

a, b Multivariate/Univariate Cox regression analyses of the association between clinical factors (including the risk score) and overall survival (OS) of patients
Correlation analysis of clinical and immune phenotype using the four model genes
To explore the relationship between the four model genes and clinical and immunophenotypes, the TCGA-UM cohort was divided into high- and low-expression groups according to the expression levels of these genes, and survival analysis was performed to examine their relevance to clinical outcomes. The patients in the high-expression groups for PTPN12 (TCGA: p = 0.029), IDH2 (TCGA: p = 0.013), P2RX4 (TCGA: p < 0.001), and KDELR2 (TCGA: p < 0.001) showed survival risk against low expression groups (Fig. 7a–d). We also found that the expression of these genes was positively correlated with the levels of CD8+ T cells (Fig. 7e–h). Moreover, patients with low levels of CD8+ T cell infiltration had longer OS (Fig. 7i–l). To determine more specifically the relationship between these genes and clinical phenotypes and immunophenotypes, we drew multiple sets of box plots. The CD8+ T cell infiltration level and immune score in the high-expression groups of these four genes were higher than those in the low-expression groups. In contrast, tumor purity was lower in the low-expression groups (Fig. 8a–c). These results suggest that the expression of the four genes may lead to poor prognosis by promoting CD8+ T cell infiltration.

Relationship of model genes with OS, CD8+ T cell levels, and TME. a–d Survival analysis using PTPN12, IDH2, P2RX4, and KDELR2. e–h The correlation of PTPN12, IDH2, P2RX4, and KDELR2 expression with CD8+ T cell levels. i–l Survival analysis using the CD8 + T cell level, immune score, ESTIMATE score, and tumor purity

a Phenotype analysis of modular genes with CD8+ T cell enrichment score. b Phenotype analysis of modular genes with immune score. c Phenotype analysis of modular genes with tumor purity
GSEA
The results of GSEA showed that high PTPN12, IDH2, P2RX4, and KDELR2 expression was significantly related to immune-related pathways such as antigen processing and presentation, chemokine signaling, cytokine-cytokine receptor interaction, T cell receptor signaling, and B cell receptor signaling (Fig. 9a-d). These pathways are also involved in tumor immunity by protecting against tumor infiltration, thus providing a reference for exploring the mechanism whereby hub genes lead to poor prognosis by promoting CD8+ T cell infiltration.

GSEA: a–d The results of immune-related GSEA analysis
Pan-cancer analysis
The above results demonstrated the role of PTPN12, IDH2, P2RX4, and KDELR2 in UM. To explore whether PTPN12, IDH2, P2RX4, and KDELR2 can promote CD8+ T cell infiltration in other types of tumors, TIMER2.0 was used to analyze their correlation. As shown in Fig. 10, these four genes were also related to the level of CD8+ T cell infiltration in some tumors. Among thirty-six tumor types excluding uveal melanoma, PTPN12 was associated with CD8+ T cell infiltration in eight tumors including bladder urothelial carcinoma (BLCA), cholangio carcinoma (CHOL), and lymphoid neoplasm diffuse large B-cell lymphoma (DLBC); KDELR2 was associated with CD8+ T cell infiltration in four tumors including DLBC, rectum adenocarcinoma (READ), and sarcoma (SARC); P2RX4 was associated with CD8+ T cell infiltration in adrenocortical carcinoma (ACC) and skin cutaneous melanoma (SKCM); IDH2 was only associated with CD8+ T cell infiltration in thymoma (THYM).

Pan-cancer analysis. a–o Pan-cancer analysis results of PTPN12, IDH2, P2RX4, and KDELR2, and correlation analysis of KICH (Kidney Chromophobe), TGCT (Testicular Germ Cell Tumors), BLCA (Bladder Urothelial Carcinoma), PCPG (Pheochromocytoma and Paraganglioma), READ (Rectum adenocarcinoma), CHOL (Cholangio carcinoma), DLBC (Lymphoid Neoplasm Diffuse Large B-cell Lymphoma), THCA (Thyroid carcinoma), PRAD (Prostate adenocarcinoma), SARC (Sarcoma), ACC (Adrenocortical carcinoma), SKCM (Skin Cutaneous Melanoma), THYM (Thymoma), and CD8+ T cells
Discussion
Owing to its high mortality, UM has brought enormous socioeconomic pressure to patients and their families. Immune checkpoint inhibitors (ICIs) offer a new therapeutic strategy, ICIs are usually antibodies that block proteins expressed on specific immune system cells (especially T cells) and tumor cells. These antibodies provide checkpoints that help keep the immune response from becoming too strong. Blocking these checkpoints allows T cells to be more powerful in killing cancer cells. The most widely used targets of ICIs are CTLA-4 and PD-1 [13]. Although these antibodies are effective in treating cutaneous melanoma (CM), they are less effective in treating UM [5, 6]. This may be due to the fact that the eye is an immune-privileged site that limits the onset of immune-mediated inflammation. Furthermore, the lack of lymphatic drainage in the eye allows the retention of tumor antigens in the ocular environment, ultimately leading to CD8+ T cell exhaustion because of continuous exposure [15, 35]. In addition, the mutational burden of UM is particularly low, unlike CM, which is known for its very high mutational burden, mostly because the skin is exposed to ultraviolet radiation, which may also account for the limited efficacy of ICIs against UM [36]. Although many immunotherapies have not achieved satisfactory efficacy in metastatic UM, with the advent of tebentafusp and improved awareness of the tumor microenvironment by researchers, immunotherapy continues to show great potential for UM.
In this study, we sought to identify co-expressed prognostic related genes that promote CD8+ T cell infiltration, which may help reveal the biological processes most closely related to CD8+ T cell infiltration. After using ssGSEA and ESTIMATE to evaluate the level of CD8+ T cell infiltration and TME in each tumor sample, WGCNA was used to construct a CD8+ T cell-related co-expression network and identity the hub module. Enrichment analysis showed that the genes in this module are mainly related to antigen processing and presentation. Moreover, a prognostic hazard model consisting of four genes (PTPN12, IDH2, P2RX4, and KDELR2) was constructed based on prognostic-related genes. These genes showed satisfactory clinical phenotype and immunophenotype correlation. Interestingly, all these genes are associated with higher CD8+ T cell infiltration and tumor progression. Finally, we evaluated the association between these four genes expression and CD8+ T cell infiltration in the other thirty-six kinds of tumors by pan-cancer analysis and found that PTPN12, KDELR2, P2RX4 and IDH2 were associated with CD8+ T cell infiltration in eight, four, two and one tumor types, respectively.
PTP-PEST is an 88 kDa protein tyrosine phosphatase (PTP) encoded by PTPN12 and containing a C-terminal PEST motif [37]. PTP-PEST serves as a protein-protein interaction domain that may regulate the intracellular half-life of proteins. Harris et al. showed that PTPN12 protect tumor cells from abnormal reactive oxygen species accumulation and oxidative stress-induced death, thereby facilitating tumor cell development [38]. A study conducted by Weidemann et al. also demonstrated that elevated PTPN12 expression parallel the tumor development and progression of prostate cancer [39]. Simultaneously, the expression of PTPN12 in immune cells is higher than that in non-immune cells [40]. In PTPN12 overexpressed-ovalbumin (OVA)-specific CD4+ T cells, PTPN12 inhibits actin reorganization and immunological synapse formation through the dephosphorylation of WASP and Arp2/3 associated signaling proteins [41]. PTPN12 also downregulates TCR-mediated activation of CD4+ and CD8+ T cells in primary human T cells and Jurkat cells, leading to decreased production of interleukin-2, production and inhibition of nuclear factor of activated T cells, nuclear factor kappa B [42]. In addition, downregulation of PTPN12 leads to the enhanced responsiveness of secondarily activated T cells including effector and memory T cells [42]. This contradicts our finding that PTPN12 promotes CD8+ T cell infiltration in UM, further suggesting that the role of infiltrating CD8+ T cells in UM may differ dramatically from other tumors. Therefore, it becomes particularly important to further explore the unique mechanism of PTPN12 action on T cells and their signature cytokines in UM, and PTP-PEST inhibitors would be a promising candidate for UM immunotherapy (Fig. 11a).

a-d Signaling pathways may associated with CD8+ T cell and Treg infiltration by four prognostic model genes, PTPN12, IDH2, P2RX4, and KDELR2.
IDH2 encodes an isocitrate dehydrogenase (IDH) isoforms that is a key metabolic enzyme catalyzing the interconversion of isocitrate to α-ketoglutarate (α-KG). Frequent mutations of IDH1 and IDH2 have been found in various tumors, such as acute myeloid leukemia (AML), glioma, and chondrosarcoma [43,44,45]. Tumor-associated IDH1 and IDH2 mutations occur almost exclusively at different arginine residues in the active enzyme site [46]. The mutation site of the IDH1 gene is located at R132 and that of the IDH2 gene at R140 and R172 [47]. The IDH mutation results in a decrease in its normal enzyme catalytic activity and a corresponding decrease in the production of the reactants NADPH and α-KG [47]. However, it acquires the new enzymatic activity that catalyzed the irreversible conversion of NADPH and α-KG into D-2-hydroxyglutarate (D-2-HG), which can competitively inhibit α-KG-dependent enzymes and subsequently induce cellular metabolic reprogramming, inhibit cell differentiation, and lead to cell tumorigenesis [47]. In addition, D-2-HG triggers hypoxia-inducible transcription factor 1 (HIF-1α) protein destabilization resulting in metabolic skewing towards oxidative phosphorylation, increased Treg frequency, and reduced T helper 17 (Th17) polarization [48]. The reduction in α-KG leads to DNA hypermethylation and reduced chromatin accessibility, which limits Treg-related functional gene expression and repressive activity [49]. Under hypoxic environments, HIF-1α promotes the migration of CD8+ T cells into tumor tissues [50, 51], and its activation leads to an increase in lactate dehydrogenase A, which converts α-KG to L-2-HG [52, 53]. Increased L-2-HG levels change the gene expression and differentiation pathway of CD8+ T cells and enhance the anti-tumor ability [54] (Fig. 11b). Therefore, inhibition of IDH mutation and promotion of α-KG conversion can reduce Treg percentage and thus inhibit tumor survival.
The protein encoded by KDEL2R, a member of the KDEL (lys-aspp-glu-leu) receptor (KDELR) family, is mainly responsible for extracting soluble endoplasmic reticulum proteins from the Golgi complex to maintain dynamic equilibrium transport [55]. In addition, it plays a key role in cell secretory trafficking, cell growth, autophagy, immune response, and tumorigenesis [56,57,58,59,60]. The regulation of Golgi secretion by KDEL2R is necessary for cell invasion and metastasis, and KDEL2R inhibition can reduce lung cancer metastasis [61]. Furthermore, KDEL2R promotes the occurrence of glioblastoma by regulating the phosphorylation level of mTOR (Ser2448). And KDEL2R could activate the mTORC1 pathway associated with cell proliferation and invasiveness [62]. The study of Sun et al. demonstrated that inhibition of mTOR during T cell activation promotes the generation of central memory Tregs in mice, these central memory Tregs possess enhanced spare respiratory capacity, similar to CD8+ memory T cells, the generation of effector Tregs also requires mTOR (Fig. 11c) [63]. Wei et al. also found that KDEL2R accelerates the cell cycle and promotes the progression of breast cancer by binding to and stabilizing the centrosome protein POC5 [64].
The purinergic receptor P2 × 4 (P2 × 4R) is encoded by P2RX4 and belongs to the purinergic receptor family for ATP. This receptor functions as a ligand-gated ion channel with high calcium permeability. Huo et al. found that P2 × 4R depletion can inhibit tumor cell survival and proliferation and promote cell apoptosis [65]. Brain derived neurotrophic factor (BDNF) is a member of the neurotrophin superfamily, BDNF and its receptor TrkB plays a role in a variety of cancers [66,67,68]. In patients with stroke, the patient who had the high levels of CD8+ BDNF+ T cells had the highest levels of BDNF+ Tregs [69]. Therefore, perhaps by silencing P2 × 4R, the expression of BDNF/TrkB can be blocked and the proportion of Treg reduced, thus inhibiting the growth of UM tumor cells (Fig. 11d).
Unlike in other tumors, we found that high CD8+ T cell infiltration is a poor prognostic marker in UM. Previous studies suggested that tumor-infiltrating CD8+ T cells may function as Tregs, an important factor contributing to ocular immune privilege, which is the result of anterior chamber-associated immune deviation (ACAID) when antigens are introduced into the anterior chamber. This induces the appearance of CD8+ Tregs to inhibit Th1- and Th2-related immune responses [70, 71]. In addition, CD8+ Tregs can inhibit immune responses directly by killing immune cells, or indirectly by producing immunosuppressive molecules such as TGFβ and indoleamine 2,3-dioxygenase (INDO) [72]. Thus, CD8+ Tregs in UM promote tumor evasion of immune responses and lead to a poor prognosis. The intra tumoral/peritumoral ratio of CD8+ Granzyme B+ cells and density of intra-tumoral CD8+ CTLs is higher in patients with disease control than in those with progressive disease, revealing the importance of elevating tumor-killing T cells in UM. In our study, four genes associated with CD8+ T cell and Treg infiltration were identified, as shown in Fig. 11, regulating the activation or suppression of CD8+ T cells and Treg through novel signaling pathways. Based on these newly identified genes and biological processes, which could contribute to the development of new UM immunotherapeutic agents.
Our study demonstrated the critical role of CD8+ T cell-mediated immune infiltration in the poor prognosis of uveal melanoma. Besides, Skalet et al. identified a population of circulating tumor cells known as circulating hybrid cells (CHCs) in the peripheral blood of uveal melanoma patients, and this hybrid cell, which combines the characteristics of both tumor cells and macrophages, may play an indispensable role in tumor progression and metastasis, with the potential to serve as a non-invasive biomarker for uveal melanoma. CHC that diffuse into the circulation provide a suitable environment for the development of primary tumors, thus driving the metastatic process. The study by Skalet et al. also identified HTR2B as an enhanced CHC detection marker to identify high-risk uveal melanomas in combination with gp100 for improved detection [73]. Additionally, the presence of dual nature cells (DNC) with tumor and leukocytic cell features, which may correspond to CHC in the blood, was identified in UM samples. The investigators found that DNC were present in 63% of UM cases and that inflammation, which represents a poorer prognosis for patients, was present in these cases [74]. Our study identified that PTPN12, IDH2, P2RX4, and KDELR2 can influence the prognosis of UM by prompting the CD8+ T cell infiltration. Further exploration of the effects of these four genes on hybrid cells may help to further reveal the secrets of UM pathogenesis. In addition, both hybrid cells and CD8+ T cells are significant prognostic markers for UM, so it is necessary to explore the interaction between these two types of cells and tumor cells.
Overall, the effect of PTPN12, IDH2, P2RX4, and KDELR2 on tumor immune cell infiltration is a complex process that typically involves multiple molecular pathways and signaling pathways. The tyrosine phosphatase encoded by PTPN12 is involved in signal transduction pathways that regulate immune cell activation and infiltration [75]. Variants of PTPN12 in tumors may affect the recognition and infiltration capacity of immune cells and thus the infiltration of CD8+ T cells. Variations in KDELR2 may indirectly affect CD8+ T cell infiltration by affecting intracellular protein transport and metabolic pathways [57]. The protein encoded by P2RX4 plays an important role in immune cell activation and infiltration as a member of the ATP-gated channel family [76]. Variations in P2RX4 may lead to aberrant activation or inhibition of signaling pathways, which may affect CD8+ T cell infiltration. Variations in IDH2, which encodes an IDH involved in cellular energy metabolism and oxidative stress responses, may affect cellular metabolic and immunoregulatory pathways, leading to alterations in the tumor microenvironment, which in turn affects CD8+ T cell infiltration [77, 78]. Aberrant gene expression or mutation also affects the immune response, tumor microenvironment, and immune escape mechanisms of the tumor, which influence immune cell infiltration. Different tumor types have different gene variants and expression profiles, and therefore, as shown by the results of our pan-cancer analyses, these genes are only associated with CD8+ T cell infiltration in specific types of tumors, but not in all tumors. The effect of these genes on CD8+ T cell infiltration in tumors should be judged depending on factors such as tumor type, genetic mutation profile, tumor microenvironment, and patient characteristics. More in-depth research and analysis are still needed in the future to understand the correlation between these genes and tumor CD8+ T cell infiltration.
This article has some limitations. First, we used the TCGA-UM, GSE44295, and GSE84976 datasets for joint analysis. Although this is all the UM cohort we could find that contained prognostic information, more external cohorts need to be added for cross-validation. Second, we hypothesized that these genes can lead to poor immunotherapy efficacy by promoting the infiltration of CD8+ T lymphocytes; however, the efficacy of immunotherapy was not included in the TCGA-UM follow-up data and therefore, more immunotherapy follow-up data need to be added. Finally, we only discussed the role of these genes in tumor development, and the specific mechanism needs further study.
In conclusion, we constructed and verified a prognostic hazard model composed of the four genes, PTPN12, IDH2, P2RX4, and KDELR2, based on a CD8+ T cell infiltration co-expression network. These four co-expressed genes mainly promote CD8+ T cell infiltration by enhancing antigen processing and presentation, and their expression leads to a poor prognosis. This study can potentially provide novel biomarker and therapeutic targets for UM.
Availability of data and materials
The datasets analyzed during the current study are available in TCGA database (The Cancer Genome Atlas, http://cancergenome.nih.gov/), and GEO database (Gene Expression Omnibus database, https://www.ncbi.nlm.nih.gov/geo/).
References
Jager MJ, Shields CL, Cebulla CM, Abdel-Rahman MH, Grossniklaus HE, Stern MH, Carvajal RD, Belfort RN, Jia R, Shields JA, et al. Uveal melanoma. Nat Rev Dis Primers. 2020;6(1):24.
Ponti A, Denys A, Digklia A, Schaefer N, Hocquelet A, Knebel JF, Michielin O, Dromain C, Duran R. First-line selective internal radiation therapy in patients with uveal melanoma metastatic to the liver. J Nuclear Med. 2020;61(3):350–6.
Killock D. Tebentafusp for uveal melanoma. Nat Rev Clin Oncol. 2021;18(12):747.
Souto EB, Zielinska A, Luis M, Carbone C, Martins-Gomes C, Souto SB, Silva AM. Uveal melanoma: physiopathology and new in situ-specific therapies. Cancer Chemother Pharmacol. 2019;84(1):15–32.
Heppt MV, Heinzerling L, Kähler KC, Forschner A, Kirchberger MC, Loquai C, Meissner M, Meier F, Terheyden P, Schell B, et al. Prognostic factors and outcomes in metastatic uveal melanoma treated with programmed cell death-1 or combined PD-1/cytotoxic T-lymphocyte antigen-4 inhibition. Eur J cancer (Oxford England: 1990). 2017;82:56–65.
Algazi AP, Tsai KK, Shoushtari AN, Munhoz RR, Eroglu Z, Piulats JM, Ott PA, Johnson DB, Hwang J, Daud AI, et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer. 2016;122(21):3344–53.
Chattopadhyay C, Kim DW, Gombos DS, Oba J, Qin Y, Williams MD, Esmaeli B, Grimm EA, Wargo JA, Woodman SE, et al. Uveal melanoma: from diagnosis to treatment and the science in between. Cancer. 2016;122(15):2299–312.
Rantala ES, Hernberg M, Kivelä TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res. 2019;29(6):561–8.
de Miguel M, Calvo E. T cell engagers in solid tumors kick the door down. Cancer Cell. 2021;39(11):1461–3.
TCR Bispecific Boosts. Survival in Uveal Melanoma. Cancer Discov. 2021;11(6):1312.
Nathan P, Hassel JC, Rutkowski P, Baurain JF, Butler MO, Schlaak M, Sullivan RJ, Ochsenreither S, Dummer R, Kirkwood JM, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385(13):1196–206.
Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591–6.
Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–21.
Meecham WJ, Char DH, Kaleta-Michaels S. Infiltrating lymphocytes and antigen expression in uveal melanoma. Ophthalmic Res. 1992;24(1):20–6.
Bronkhorst IH, Vu TH, Jordanova ES, Luyten GP, Burg SH, Jager MJ. Different subsets of tumor-infiltrating lymphocytes correlate with macrophage influx and monosomy 3 in uveal melanoma. Invest Ophthalmol Vis Sci. 2012;53(9):5370–8.
de Waard-Siebinga I, Hilders CG, Hansen BE, van Delft JL, Jager MJ. HLA expression and tumor-infiltrating immune cells in uveal melanoma. Graefes Arch Clin Exp Ophthalmol. 1996;234(1):34–42.
Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front Immunol. 2018;9:14.
Huh JW, Lee JH, Kim HR. Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch Surg. 2012;147(4):366–72.
Thomas NE, Busam KJ, From L, Kricker A, Armstrong BK, Anton-Culver H, Gruber SB, Gallagher RP, Zanetti R, Rosso S, et al. Tumor-infiltrating lymphocyte grade in primary melanomas is independently associated with melanoma-specific survival in the population-based genes, environment and melanoma study. J Clin Oncol. 2013;31(33):4252–9.
Zeng DQ, Yu YF, Ou QY, Li XY, Zhong RZ, Xie CM, Hu QG. Prognostic and predictive value of tumor-infiltrating lymphocytes for clinical therapeutic research in patients with non-small cell lung cancer. Oncotarget. 2016;7(12):13765–81.
Wang Y, Xu Y, Dai X, Lin X, Shan Y, Ye J. The prognostic landscape of adaptive immune resistance signatures and infiltrating immune cells in the tumor microenvironment of uveal melanoma. Exp Eye Res. 2020;196:108069.
Sun Y, Wu J, Yuan Y, Lu Y, Luo M, Lin L, Ma S. Construction of a promising tumor-infiltrating CD8 + T cells gene signature to improve prediction of the prognosis and immune response of uveal melanoma. Front cell Dev biology. 2021;9:673838.
Zoroquiain P, Esposito E, Logan P, Aldrees S, Dias AB, Mansure JJ, Santapau D, Garcia C, Saornil MA, Belfort Neto R, et al. Programmed cell death ligand-1 expression in tumor and immune cells is associated with better patient outcome and decreased tumor-infiltrating lymphocytes in uveal melanoma. Mod Pathology. 2018;31(8):1201–10.
Singh L, Singh MK, Kenney MC, Jager MJ, Rizvi MA, Meel R, Lomi N, Bakhshi S, Sen S, Kashyap S. Prognostic significance of PD-1/PD-L1 expression in uveal melanoma: correlation with tumor-infiltrating lymphocytes and clinicopathological parameters. Cancer Immunol Immunother. 2021;70(5):1291–303.
Al-Shibli KI, Donnem T, Al-Saad S, Persson M, Bremnes RM, Busund LT. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res. 2008;14(16):5220–7.
Koch CM, Chiu SF, Akbarpour M, Bharat A, Ridge KM, Bartom ET, Winter DR. A beginner’s guide to analysis of RNA sequencing data. Am J Respir Cell Mol Biol. 2018;59(2):145–57.
Chen JW, Shrestha L, Green G, Leier A, Marquez-Lago TT. The hitchhikers’ guide to RNA sequencing and functional analysis. Brief Bioinform. 2023;24(1):bbac529.
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Liu Y, Gu HY, Zhu J, Niu YM, Zhang C, Guo GL. Identification of hub genes and key pathways associated with bipolar disorder based on weighted gene co-expression network analysis. Front Physiol. 2019;10:1081.
Chen H, Yang J, Wu W. Seven key hub genes identified by gene co-expression network in cutaneous squamous cell carcinoma. BMC Cancer. 2021;21(1):852.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607-d613.
Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303.
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509-w514.
Rossi E, Schinzari G, Zizzari IG, Maiorano BA, Pagliara MM, Sammarco MG, Fiorentino V, Petrone G, Cassano A, Rindi G, et al. Immunological backbone of uveal melanoma: is there a rationale for immunotherapy? Cancers. 2019;11(8):1055.
Rodrigues M, Mobuchon L, Houy A, Alsafadi S, Baulande S, Mariani O, Marande B, Ait Rais K, Van der Kooij MK, Kapiteijn E, et al. Evolutionary routes in metastatic uveal melanomas depend on MBD4 alterations. Clin Cancer Res. 2019;25(18):5513–24.
Takekawa M, Itoh F, Hinoda Y, Arimura Y, Toyota M, Sekiya M, Adachi M, Imai K, Yachi A. Cloning and characterization of a human cDNA encoding a novel putative cytoplasmic protein-tyrosine-phosphatase. Biochem Biophys Res Commun. 1992;189(2):1223–30.
Harris IS, Blaser H, Moreno J, Treloar AE, Gorrini C, Sasaki M, Mason JM, Knobbe CB, Rufini A, Hallé M, et al. PTPN12 promotes resistance to oxidative stress and supports tumorigenesis by regulating FOXO signaling. Oncogene. 2014;33(8):1047–54.
Weidemann SA, Sauer C, Luebke AM, Möller-Koop C, Steurer S, Hube-Magg C, Büscheck F, Höflmayer D, Tsourlakis MC, Clauditz TS, et al. High-level expression of protein tyrosine phosphatase non-receptor 12 is a strong and independent predictor of poor prognosis in prostate cancer. BMC Cancer. 2019;19(1):944.
Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol. 2012;13(5):439–47.
Côté JF, Chung PL, Théberge JF, Hallé M, Spencer S, Lasky LA, Tremblay ML. PSTPIP is a substrate of PTP-PEST and serves as a scaffold guiding PTP-PEST toward a specific dephosphorylation of WASP. J Biol Chem. 2002;277(4):2973–86.
Davidson D, Shi X, Zhong MC, Rhee I, Veillette A. The phosphatase PTP-PEST promotes secondary T cell responses by dephosphorylating the protein tyrosine kinase Pyk2. Immunity. 2010;33(2):167–80.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Sci (New York NY). 2008;321(5897):1807–12.
Kunadt D, Stasik S, Metzeler KH, Röllig C, Schliemann C, Greif PA, Spiekermann K, Rothenberg-Thurley M, Krug U, Braess J, et al. Impact of IDH1 and IDH2 mutational subgroups in AML patients after allogeneic stem cell transplantation. J Hematol Oncol. 2022;15(1):126.
Zhu GG, Nafa K, Agaram N, Zehir A, Benayed R, Sadowska J, Borsu L, Kelly C, Tap WD, Fabbri N, et al. Genomic profiling identifies Association of IDH1/IDH2 mutation with longer relapse-free and metastasis-free survival in high-grade chondrosarcoma. Clin Cancer Res. 2020;26(2):419–27.
Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in Cancer. Cancer Cell. 2018;34(2):186–95.
Guo J, Zhang R, Yang Z, Duan Z, Yin D, Zhou Y. Biological roles and therapeutic applications of IDH2 mutations in human cancer. Front Oncol. 2021;11:644857.
Böttcher M, Renner K, Berger R, Mentz K, Thomas S, Cardenas-Conejo ZE, Dettmer K, Oefner PJ, Mackensen A, Kreutz M, et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology. 2018;7(7):e1445454.
Liu Q, Zhu F, Liu X, Lu Y, Yao K, Tian N, Tong L, Figge DA, Wang X, Han Y, et al. Non-oxidative pentose phosphate pathway controls regulatory T cell function by integrating metabolism and epigenetics. Nat Metabol. 2022;4(5):559–74.
Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8 + T cells. J Exp Med. 2012;209(13):2441–53.
Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14(11):1173–82.
Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Veliça P, You J, Chia GS, Sim J, Doedens A, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature. 2016;540(7632):236–41.
Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, et al. Hypoxia induces production of L-2-Hydroxyglutarate. Cell Metabol. 2015;22(2):304–11.
Ye D, Guan KL, Xiong Y. Metabolism, activity, and targeting of D- and L-2-Hydroxyglutarates. Trends Cancer. 2018;4(2):151–65.
Capitani M, Sallese M. The KDEL receptor: new functions for an old protein. FEBS Lett. 2009;583(23):3863–71.
Solis GP, Bilousov O, Koval A, Lüchtenborg AM, Lin C, Katanaev VL. Golgi-resident Gαo promotes protrusive membrane dynamics. Cell. 2017;170(6):1258.
Trychta KA, Bäck S, Henderson MJ, Harvey BK. KDEL receptors are differentially regulated to maintain the ER proteome under calcium deficiency. Cell Rep. 2018;25(7):1829-1840e1826.
Wang P, Li B, Zhou L, Fei E, Wang G. The KDEL receptor induces autophagy to promote the clearance of neurodegenerative disease-related proteins. Neuroscience. 2011;190:43–55.
Henderson MJ, Richie CT, Airavaara M, Wang Y, Harvey BK. Mesencephalic astrocyte-derived neurotrophic factor (MANF) secretion and cell surface binding are modulated by KDEL receptors. J Biol Chem. 2013;288(6):4209–25.
Kamimura D, Katsunuma K, Arima Y, Atsumi T, Jiang JJ, Bando H, Meng J, Sabharwal L, Stofkova A, Nishikawa N, et al. KDEL receptor 1 regulates T-cell homeostasis via PP1 that is a key phosphatase for ISR. Nat Commun. 2015;6:7474.
Bajaj R, Kundu ST, Grzeskowiak CL, Fradette JJ, Scott KL, Creighton CJ, Gibbons DL. IMPAD1 and KDELR2 drive invasion and metastasis by enhancing golgi-mediated secretion. Oncogene. 2020;39(37):5979–94.
Liao Z, She C, Ma L, Sun Z, Li P, Zhang X, Wang P, Li W. KDELR2 promotes glioblastoma tumorigenesis targeted by HIF1a via mTOR signaling pathway. Cell Mol Neurobiol. 2019;39(8):1207–15.
Sun IH, Oh MH, Zhao L, Patel CH, Arwood ML, Xu W, Tam AJ, Blosser RL, Wen J, Powell JD. mTOR complex 1 signaling regulates the generation and function of Central and Effector Foxp3(+) Regulatory T cells. J Immunol. 2018;201(2):481–92.
Wei H, Ma W, Lu X, Liu H, Lin K, Wang Y, Ye Z, Sun L, Huang Z, Pan T, et al. KDELR2 promotes breast cancer proliferation via HDAC3-mediated cell cycle progression. Cancer Commun. 2021;41(9):904–20.
Huo JF, Chen XB. P2 × 4R silence suppresses glioma cell growth through BDNF/TrkB/ATF4 signaling pathway. J Cell Biochem. 2019;120(4):6322–9.
Ding D, Hou R, Gao Y, Feng Y. miR-613 inhibits gastric cancer progression through repressing brain derived neurotrophic factor. Exp Ther Med. 2018;15(2):1735–41.
Yuan Y, Ye HQ, Ren QC. Upregulation of the BDNF/TrKB pathway promotes epithelial-mesenchymal transition, as well as the migration and invasion of cervical cancer. Int J Oncol. 2018;52(2):461–72.
Jóźwiak-Bębenista M, Jasińska-Stroschein M, Kowalczyk E. The differential effects of neuroleptic drugs and PACAP on the expression of BDNF mRNA and protein in a human glioblastoma cell line. Acta Neurobiol Exp. 2017;77(3):205–13.
Chan A, Yan J, Csurhes P, Greer J, McCombe P. Circulating brain derived neurotrophic factor (BDNF) and frequency of BDNF positive T cells in peripheral blood in human ischemic stroke: Effect on outcome. J Neuroimmunol. 2015;286:42–7.
Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006;7(4):354–9.
Niederkorn JY. Emerging concepts in CD8(+) T regulatory cells. Curr Opin Immunol. 2008;20(3):327–31.
Vlad G, Suciu-Foca N. Resurgence or emergence of CD8 + ts. Hum Immunol. 2008;69(11):679–80.
Parappilly MS, Chin Y, Whalen RM, Anderson AN, Robinson TS, Strgar L, Sutton TL, Conley P, Klocke C, Gibbs SL, et al. Circulating neoplastic-immune hybrid cells predict metastatic progression in uveal melanoma. Cancers. 2022;14(19):4617.
Marcotte E, Lopez D, Goyeneche AA, Abdouh M, Youhnovska EN, Bergeron S, Dias AB, Burnier MN. Discovering the pathogenesis of cancer dissemination: the role of dual nature – hybrid cells in uveal melanoma. Investig Ophthalmol Vis Sci. 2023;64(8):910–910.
Lee C, Rhee I. Important roles of protein tyrosine phosphatase PTPN12 in tumor progression. Pharmacol Res. 2019;144:73–8.
Kanellopoulos JM, Almeida-da-Silva CLC, Rüütel Boudinot S, Ojcius DM. Structural and functional features of the P2 × 4 receptor: an immunological perspective. Front Immunol. 2021;12:645834.
Lee JH, Go Y, Kim DY, Lee SH, Kim OH, Jeon YH, Kwon TK, Bae JH, Song DK, Rhyu IJ, et al. Isocitrate dehydrogenase 2 protects mice from high-fat diet-induced metabolic stress by limiting oxidative damage to the mitochondria from brown adipose tissue. Exp Mol Med. 2020;52(2):238–52.
Stein EM. Molecular pathways: IDH2 mutations-co-opting cellular metabolism for malignant transformation. Clin Cancer Res. 2016;22(1):16–9.
Acknowledgements
We acknowledge TCGA and GEO database for providing their platforms and contributors for uploading their meaningful datasets.
Funding
Clinical Research Incubation Project, West China Hospital, Sichuan University (2021HXFH026); Chengdu Municipal Science and Technology Bureau Key R&D Support Program (2021-YF09-00024-SN); Sichuan Provincial Science and Technology Department Key R&D Projects (2023YFQ0103).
Author information
Authors and Affiliations
Contributions
CZ, JX, and ZX designed and supervised the study. CZ, LF, FJ, and HJ analyzed the data. CZ and JX wrote the original draft. LZ and ZX edited the draft. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
TCGA and GEO are public databases. The patients involved in the database have obtained ethical approval. Users can download relevant data for free to research and publish related articles. Our research is based on open source data, so there are no ethical issues or other conflicts of interest.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, C., Xiao, J., Fa, L. et al. Identification of co-expressed gene networks promoting CD8+ T cell infiltration and having prognostic value in uveal melanoma. BMC Ophthalmol 23, 354 (2023). https://doi.org/10.1186/s12886-023-03098-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-023-03098-7