
Abstract
Background
Usher syndrome (USH) is an autosomal recessive disorder primarily responsible for deaf-blindness. Patients with subtype Usher syndrome type 1 (USH1) typically experience congenital sensorineural hearing loss, abnormal vestibular function, and retinitis pigmentosa (RP). Here we present a case of Usher syndrome type 1F (USH1F) with a novel homozygous variant in the calcium-dependent cell-cell adhesion protocadherin-15 (PCDH15) gene.
Case presentation
Ophthalmic examinations were evaluated over a course of 10 years and the disease-causing variant was identified by whole exome sequencing (WES). Initial and follow-up examination of color fundus photos after 10 years revealed an increase in bone spicule pigment deposits in both eyes. A parafoveal hyper-AF ring in both eyes was shown in fundus autofluorescence (FAF) with a progressive diameter-wise constriction observed over 8 years. Outer nuclear layer (ONL) loss was observed in parafoveal and perifoveal regions of both eyes on spectral domain–optical coherence tomography (SD-OCT). Full-field electroretinography (ffERG) showed extinguished global retinal function. WES identified a novel two-base-pair deletion, c.60_61del (p.Phe21Ter), in the PCDH15 gene, confirming the diagnosis of USH1F.
Conclusions
We report a novel homozygous PCDH15 pathogenic variant expected to lead to nonsense-mediated decay (NMD) of PCDH15 mRNA. The patient exhibits a loss of function with USH1F, experiencing congenital hearing loss and syndromic RP.
Similar content being viewed by others

Background
Usher syndrome (USH) is an autosomal recessive disorder that is widely responsible for deaf-blindness. This genetic disorder was first reported in 1914 by Charles Usher, who described 69 patients from 40 separate families presenting with retinitis pigmentosa and hearing loss (Usher, 1914). Usher syndrome is a ciliopathy that disrupts photoreceptor ciliogenesis in the retina and kinocilia in the inner ear [1]. Patients with USH are characterized by rod-cone dystrophy, partial-complete sensorineural hearing loss, and a possibility of vestibular dysfunction [2]. The severity and onset of symptoms depend on the clinical type — Usher type 1 (USH1), attributed to variants in protein coding genes USH1C, CDH23, PCDH15, and USH1G; Usher type 2 (USH2) from variants in USH2A, ADGRV1, and WHRN; Usher type 3 (USH3) from CLRN1 [3]; and atypical Usher syndrome type 4 (USH4) from ARSG [4].
Variants in PCDH15 are responsible for 11–19% of USH1 cases and is categorized specifically as Usher type 1F (USH1F, OMIM: 602083). USH1F is typically described by congenital sensorineural hearing loss, abnormal vestibular function, and the prepubertal onset of progressive retinitis pigmentosa (RP) [2]. The PCDH15 gene spans a genomic region of 980 kb composed of 33 exons. It encodes for the protocadherin-15 protein, belonging to integral membrane proteins that mediate calcium-dependent cell-adhesion, which play a crucial role in retinal and cochlear function [3].
In this report, we present a case of USH1F with a novel homozygous variant c.60_61del in the PCDH15 gene (NM_001384140.1), creating a stop codon (p.Phe21Ter) after the 20th amino acid.
Case presentation
A 21-year-old East-Asian female presented to our clinic with peripheral vision loss and impaired night vision. Past medical history included congenital hearing loss, for which cochlear implant surgery was done in her early childhood. The patient is the product of a reportedly nonconsanguineous union between healthy parents and had no pertinent family history. The patient reported an unaffected brother and no history of smoking and drinking.
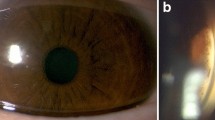
At the time of presentation, the patient displayed a best-corrected visual acuity (BCVA) of 20/50 and 20/40 in the right eye and left eye, respectively. Initial color fundus images revealed mild, bilateral retinal vessel attenuation and bone spicule pigment deposits in the mid-periphery of both eyes (Fig. 1a).

Color fundus of both eyes. a At presentation, fundus shows mild bilateral retinal vessel attenuation and bone spicule deposits in both eyes. b Follow up fundus reveals a pale retina with a progression in bilateral retinal vessel attenuation and amount of bone spicule pigment deposits
Fundus autofluorescence (FAF) images revealed an abnormal parafoveal hyperautofluorescence (hyper-AF) ring, surrounded by hypo-AF spots in both eyes (see Table 1 and Fig. 2a). Initial spectral domain-optical coherence tomography (SD-OCT) images revealed the preservation of the external limiting membrane (ELM) and inner segment ellipsoid (ISe) band in only the foveal region. Significantly reduced outer nuclear layer (ONL) thickness in the parafoveal and perifoveal area suggested heavy photoreceptor degeneration (Fig. 2b). Cystoid macular edema was observed in the inner nuclear layer (Fig. 2b). The aligned FAF and SD-OCT images revealed that the outer border of the hyper-AF ring aligns with the point of ELM disruption. These observations correspond with previous assessments of hyper-AF ring structure in patients with RP [5]. Full-field electroretinogram (ffERG) tests displayed an extinguished rod response, combined rod-cone response, cone response, and 30 Hz flicker response in both eyes since the time of presentation (Supplementary Fig. 1).

Initial fundus autofluorescence (FAF) and spectral domain optical coherence tomography (SD-OCT) of both eyes. a FAF at presentation reveals a parafoveal hyper-AF ring and in both eyes. b SD-OCT at presentation shows outer nuclear layer (ONL), external limiting membrane (ELM), and inner segment ellipsoid (ISe) loss in the parafoveal and perifoveal regions in both eyes. b Cystoid macular edema in right eye can be observed in parafoveal region. Follow-up fundus autofluorescence (FAF) and spectral domain optical coherence tomography (SD-OCT) images of both eyes. c Follow up FAF shows a constriction in the parafoveal hyper-AF ring diameter. d Follow up SD-OCT shows an equivalent shortening in ELM and ISe bands in both eyes with additional reduction in ONL thickness in both eyes
Follow-up examinations were conducted over a course of 10 years. At 31-years of age, the patient’s BCVA remained consistent to initial measurements at presentation (see Table 1 for details). However, the patient’s fundus image shows a progression in bilateral retinal vessel attenuation and retinal depigmentation, along with an increased amount of bone spicule pigment deposits in the mid-periphery region, extending towards the near-peripheral retina (Fig. 1b).
Follow-up FAF and SD-OCT images show a progressive diameter-wise constriction in the perifoveal hyper-AF ring (see Table 1 and Fig. 2c), with an equivalent shortening in ELM and ISe bands in both eyes (Fig. 2d). An additional reduction in ONL thickness was also observed in the follow-up SD-OCT images in both eyes (Fig. 2d).
The patient was diagnosed with Usher syndrome type 1 based on the presented characteristics of congenital hearing loss and syndromic RP. Subtype USH1F was confirmed by whole exome sequencing (WES) that revealed a previously unseen homozygous variant, c.60_61del in the PCDH15 gene (NM_001384140.1), which is predicted to create a stop codon (p.Phe21Ter) after the 20th amino acid. The variant was further confirmed by Sanger sequencing (Supplementary Fig. 2).
Discussion and conclusions
The patient presenting with RP and congenital hearing loss was monitored over a course of 10 years. Progression in RP was observed with an increase in bone spicule deposits, a constriction of the perifoveal hyper-AF ring, and a reduction in ONL thickness. Since PCDH15 is abundantly expressed in both rod and cone photoceptors [6], the patient’s clinical progression indicates significant photoreceptor degeneration associated with the novel two-base-pair deletion (c.60_61del) in PCDH15 (NM_001384140.1). This variant is predicted to produce a premature stop codon in exon 2/33, which is expected to result in nonsense-mediated decay (NMD) of PCDH15 mRNA. The impaired expression of PCDH15 in rod and cone photoreceptors is likely associated with the patient’s progression in RP with a loss in rod photoreceptors and secondary cone degeneration. Although the exact molecular function of retinal protocadherin-15 is ambiguous [7], using existing animal models with PCDH15 variants may help further understand the patient’s clinical observations. A similar founder variant in PCDH15 that is unique to Ashkenazi Jews, p.Arg245Ter, was simulated using a Pcdh15R250X knockin mutant mouse model that phenocopied human p.Arg245Ter congenital hearing loss and abnormal vestibular function [7]. While present in wild-type mice, immunostaining revealed the absence of protocadherin-15 expression in the inner segments of photoreceptors, outer plexiform layer, the ganglion cell layer, and retinal pigment epithelium (RPE) in Pcdh15R250X mice. Under photopic conditions, the loss of protocadherin-15 hinders the transportation of arrestin and transducin between the photoreceptor outer segment (OS) and inner segment (IS) to desensitize or bind to opsin, respectively. This results in abnormal protein localization in the phototransduction cascade and retinoid cycle [7]. Additionally, a reduction in enzymes CRALBP and RPE65 consequently reduced 11-cis-retinal functions [7]. Combined with the gross retinal degeneration observed in the patient, both observations may help explain extinct ffERG amplitudes in Supplementary Fig. 1. However, acute retinal degeneration was less severe in mice when compared to human pathophysiology [7]. This may be attributed to the absence of protocadherin-15 associated calyceal processes in rodent photoreceptor cells that are present in humans, frogs, and monkeys [8]. Knockdown PCDH15 frog models show the degeneration and loss of photoreceptor function due to the proposed role of the calyceal process in rod and cone maintenance and development [8].
The c.60_61del variant was identified as homozygous. Although the parents were not known to be related to each other, the patient had regions of homozygosity (ROH) across ~ 0.8% of the genome suggesting that the parents are distantly related. The PCDH15 homozygous variant was found within one of the larger ROH (~ 10 Mb). Therefore, even though the parents were not available for Sanger sequencing, it is likely that each parent is heterozygous for the variant. There was no evidence of a large copy-number-loss variant spanning the PCDH15 gene from the WES data.
Loss of function variants are known to be the disease-causing mechanism with many pathogenic variants reported in the literature and databases being null variants. Clinical observations reported a loss of function in the patient’s phenotype, which may be attributed to NMD mechanisms eliminating mRNA containing a premature termination codon [9]. Since the variant is located at the beginning of the PCDH15 gene on exon 2/33, nonsense-mediated decay of PCDH15 mRNA is expected to occur. However, if a small amount of mRNA does escape the NMD pathway, then the shortened peptide (around 21 amino acids), will most likely be degraded. Hence, it is likely that no protein products are produced. To further examine the mechanisms that relate to a loss of function, future animal models may use qPCR techniques to determine the expression of PCDH15 mRNA to examine the role of NMD in this novel variant.
In this longitudinal report, we followed a patient with a novel variant in the PCDH15 gene over a course of 10 years. A novel nonsense variant c.60_61del results in typical USH1F clinical symptoms, such as congenital hearing loss and progressive RP [2]. Although the patient was first examined in her twenties, with clinical features resembling clinical subtypes of USH 2 and USH 3 [2], her ERG responses (Supplementary Fig. 1) resemble RP in the advanced stage [10] suggesting a prepubertal onset of photoreceptor degeneration, which correspond with bilateral ONL thinning in SD-OCT (Fig. 2d). Thus, ffERG is essential for an early and correct diagnosis of USH1 when combined with the presence of congenital hearing loss, allowing clinicians to test genes related to USH1.
Availability of data and materials
The dataset that used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- USH:
-
Usher syndrome
- USH1:
-
Usher syndrome type 1
- USH2:
-
Usher syndrome type 2
- USH3:
-
Usher syndrome type 3
- USH4:
-
Usher syndrome type 4
- USH1F:
-
Usher type 1F
- BCVA:
-
Best-corrected visual acuity
- FAF:
-
Fundus autofluorescence
- SD-OCT:
-
Spectral domain-optical coherence tomography
- ELM:
-
External limiting membrane
- ISe:
-
Inner segment ellipsoid
- ONL:
-
Outer nuclear layer
- ffERG:
-
Full-field electroretinogram
- WES:
-
Whole exome sequencing
- NMD:
-
Nonsense-mediated decay
- RPE:
-
Retinal pigment epithelium
- OS:
-
Outer segment
- IS:
-
Inner segment
- ROH:
-
Regions of homozygosity
References
Fuster-Garcia C, Garcia-Bohorquez B, Rodriguez-Munoz A, Aller E, Jaijo T, Millan JM, et al. Usher syndrome: genetics of a human ciliopathy. Int J Mol Sci. 2021;22(13):6723.
Tsang SH, Aycinena ARP, Sharma T. Ciliopathy: usher syndrome. Adv Exp Med Biol. 2018;1085:167–70.
Castiglione A, Moller C. Usher syndrome. Audiol Res. 2022;12(1):42–65.
Velde HM, Reurink J, Held S, Li CHZ, Yzer S, Oostrik J, et al. Usher syndrome type IV: clinically and molecularly confirmed by novel ARSG variants. Hum Genet. 2022;141(11):1723–38.
Lima LH, Cella W, Greenstein VC, Wang NK, Busuioc M, Smith RT, et al. Structural assessment of hyperautofluorescent ring in patients with retinitis pigmentosa. Retina. 2009;29(7):1025–31.
Sun X, Pawlyk B, Adamian M, Michaud N, Bulgakov OV, Li T. Functional and structural deficits of cone photoreceptors in mice lacking PCDH15, a protein encoded by the Ush1F gene. Invest Ophthalmol Vis Sci. 2006;47(13):5770.
Sethna S, Zein WM, Riaz S, Giese AP, Schultz JM, Duncan T, et al. Proposed therapy, developed in a Pcdh15-deficient mouse, for progressive loss of vision in human usher syndrome. Elife. 2021;10:e67361.
Schietroma C, Parain K, Estivalet A, Aghaie A, Boutet de Monvel J, Picaud S, et al. Usher syndrome type 1-associated cadherins shape the photoreceptor outer segment. J Cell Biol. 2017;216(6):1849–64.
Howard MT, Malik N, Anderson CB, Voskuil JL, Atkins JF, Gibbons RJ. Attenuation of an amino-terminal premature stop codon mutation in the ATRX gene by an alternative mode of translational initiation. J Med Genet. 2004;41(12):951–6.
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86.
Acknowledgements
Not applicable.
Funding
Nan-Kai Wang and his lab is supported by the National Institute of Health R01EY031354, P30EY019007, Vagelos College of Physicians & Surgeons (VP&S) Grants and Gerstner Philanthropies. Laura Liu and her lab are supported by a Chang Gung Memorial Hospital Research Grant CMRPG3M0631. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Data collection, A.H.K., P.-K.L., E.Y.-C.K., Y.-J.T., L.L, K.-J.C., and W.-C.W.; genomic data analysis, G.H.S., H.L., and R.K.; image analysis, N.C., M.-C.H., and N.-K.W.; writing-original draft, N.C., M.-C.H., and N.-K.W.; writing-review and editing, N.C., A.H.K., P.-K. L., E.Y.-C.K., H.L., W.-C.W., M.-C.H., and N.-K.W. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patient who received genetic tests in this report had previously signed consent forms for another prospective study on “Genetic study in hereditary retinal and optic nerve diseases, (IRB201601569B0C601)”.
Consent for publication
The patient has consented for the publication of their examination results and other personal or clinical details and images. Signed consent forms are available from the corresponding author on reasonable request.
Competing interests
The authors report no conflicts of interest and are alone responsible for the content and writing of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Fig. 1.
Full-field electroretinography (ffERG) at presentation shows extinguished rod response, combined rod-cone response, cone response and 30 Hz flicker response in both eyes.
Additional file 2: Supplementary Fig. 2.
Sanger sequencing of the PCDH15 gene. The sequence trace shows the PCDH15 variant, which is consistent with whole exome sequencing (WES) test results.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, N., Lee, H., Kim, A.H. et al. Case report: novel PCDH15 variant causes usher syndrome type 1F with congenital hearing loss and syndromic retinitis pigmentosa. BMC Ophthalmol 22, 441 (2022). https://doi.org/10.1186/s12886-022-02659-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-022-02659-6