
Abstract
Background
To identify the genetic mutation of a four-generation autosomal dominant congenital cataract family in China.
Methods
Targeted region sequencing containing 778 genes associated with ocular diseases was performed to screen for the potential mutation, and Sanger sequencing was used to confirm the mutation. The homology model was constructed to identify the protein structural change, several online software were used to predict the mutation impact. CLUSTALW was used to perform multiple sequence alignment from different species.
Results
A novel heterozygous mutation, GJA8 NM_005267.5: c.124G > A, p.(E42K) was found, which cosegregated with congenital cataract phenotype in this family. Bioinformatics analysis of the mutation showed that the surface potential diagram of proteins changed. Several online programs predicted the mutation was ‘Pathogenic’, ‘Damaging’, ‘Disease causing’ or ‘Deleterious’.
Conclusions
A novel mutation NM_005267.5(GJA8):c.124G > A was identified in our study. Our finding can broaden the mutation spectrum of GJA8, enrich the phenotype-genotype correlation of congenital cataract and help to better understand the genetic background of congenital cataract.
Similar content being viewed by others

Background
Congenital cataract is defined as lens opacity that presents at birth or during the first decade of life [1]. It is the leading cause of visual impairment and reversible blindness in childhood [2]. The prevalence of congenital cataract ranges from 0.63 to 9.74 per 10 000, which varies with regional socioeconomic development [3, 4]. Although multiple factors can cause congenital cataract, genetic inheritance is the most common one [1]. Nearly one third of cases have a genetic basis, and the most frequent mode of inheritance is autosomal dominant transmission with a high degree of penetrance [5, 6].
Up to now, at least 100 genes have been identified in syndromic and nonsyndromic congenital cataract [6]. Those known mutant genes encode proteins including crystalline (CRYAA, CRYAB, CRYBA1/A3, CRYBA4, CRYBB1, CRYBB2, CRYGC, CRYGD and CRYGS), gap junction proteins (GJA3 and GJA8, also called Cx50), membrane protein (MIP/AQP0), beaded filament proteins (BFSP1 and BFSP2), growth and transcription factors (HSF4 and PITX3), and others (CHMP4B and EPHA2) [7, 8]. However, there is no clear correlation between genotype and phenotype for inherited cataract. Mutations in the same gene can result in different cataract phenotypes and mutations in different genes can lead to similar cataract phenotypes.
In the present study, we tried to identify the genetic mutation in an autosomal dominant inherited four-generation cataract family. By targeted region sequencing, a heterozygous missense mutation, GJA8 NM_005267.5: c.124G > A, p.(E42K) was found. This is a novel mutation that has not been reported previously.
Methods
Patients
A four-generation Chinese family from Tianjin was recruited in the present study. This family consists of 19 individuals, in which 3 male and 4 female are suffering from congenital cataract. All the patients of this family accepted ophthalmologic examinations and all the patients except the proband had already accepted cataract extraction and intraocular lens implantation surgery. The informed consent was obtained from all subjects of this pedigree after explanation of the nature and possible consequences of the study. This study was approved by the ethics committee of Tianjin Medical University Eye Hospital and followed the tenets of the Declaration of Helsinki. The peripheral blood samples were obtained from ten family members, including five patients and five unaffected individuals, and drawn into an ethylenediamine tetraacetic acid (EDTA) sample tube for further analysis.
DNA sequencing
The method of targeted region sequencing and data analysis was described in detail before [9, 10]. Briefly, genomic DNA was extracted according to the manufacturer’s standard procedure (MagPure Buffy Coat DNA Midi KF Kit, Magen, China). The qualified genomic DNA was sequenced with PE100 + 100 on MGISEQ-2000. The BGI MGIEasy V4 chip, containing 778 genes associated with ocular diseases through OMIM (Table 1), was used to capture the targeted sequences. Sanger sequencing was used to validate all mutations and potential pathogenic variants after PCR amplification using the primers: forward, GTGCACATTGACCGTTCTGG; reverse, CCTCCAGCCGGAACTTCTTA. Segregation analysis was performed in all available family members. The mutation was also blasted in ESP6500, ExAC, GnomAD, GnomAD-EAS, NCBI dbSNP, HapMap, 1000 human genome dataset and database of 100 Chinese healthy adults in order to rule out the possibility of a polymorphism.
Bioinformatics analysis
Using the solved structure of gap junction protein alpha 8 as template (Protein Data Bank accession No.6MHY_A), the model structure of homomeric wild and the mutant GJA8 were constructed by Swiss Model Server (https://swissmodel.expasy.org) and shown with the PyMOL Molecular Graphic system (Delano Scientific). Meanwhile, we used online programs to predict the possible functional impact of the amino acid change, including BayesDel addAF, BayesDel no AF, DANN, DEOGEN2, EIGEN, FATHMM, LRT, MVP, MetaLR, MetaSVM, MutPred, MutationTaster, PROVEAN, PrimateAI, REVEL, SIFT, etc. (http://varsome.com). In addition, multiple sequence alignment from different species was performed by CLUSTALW (https://www.genome.jp/tools-bin/clustalw).
Results
Clinical evaluation
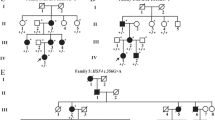
Ten individuals from this four-generation family were recruited in this study, including 5 affected individuals (I:2, II:2, II:5, III:2, IV:1) (Fig. 1a). The proband (II:2), a 46-year-old female, was diagnosed bilateral cataract when she was 3 years old. With the visual acuity decreasing gradually, her left eye underwent cataract extraction and intraocular lens implantation surgery when she was 44 years old. The lens of her right eye was observed to be like a “full moon” with central pulverulent opacities (Fig. 1b). According to the history and medical records, all the other affected individuals were diagnosed bilateral cataract since their childhood and accepted cataract surgery before. Apart from cataract, there were no other ocular or systemic abnormalities.

Clinical evaluation of a Chinese pedigree with congenital cataract. a A four-generation family with autosomal dominant congenital cataract. The arrow indicates the proband. Squares and circles symbolize males and females, respectively. Black and white denotes affected and unaffected individuals, respectively. W represents wild-type GJA8 allele; M represents allele with mutation. b Slit-lamp photograph of the proband showed pulverulent nuclear cataract
Identification of GJA8 mutations
Targeted region sequencing containing 778 genes associated with ocular diseases was performed for the proband (II:2). The average read depth was 139.02X, the sequence coverage of the targeted region was 99.96% and the percentage of the average read depth over 30X was 95.58%. After filtering, one heterozygous mutation GJA8 NM_005267.5: c.124G > A, p.(E42K) was identified, which was considered to be associated with congenital cataract. This c.124G > A nucleotide replacement causes a substitution of positively charged lysine for a negatively charged wild-type glutamic acid at codon 42 (p.E42K). The mutation was further confirmed by Sanger sequencing (Fig. 2a, b).

Sanger sequencing of GJA8. a The red arrow indicates the G>A transition at codon 42 (underlined) in an affected patient from the family. AAG encodes Lys (K). b Wild-type sequence from an unaffected family member. GAG encodes Glu (E)
Segregation analysis demonstrated that this mutation was detected in all the five affected individuals of this family and not detected in the unaffected family members (II:1, II:6, II:7, III:1, III:7), indicating the mutant NM_005267.5(GJA8):c.124G > A cosegregated with congenital cataract phenotype in this family. The mutation was not found in ESP6500, ExAC, GnomAD, GnomAD-EAS, NCBI dbSNP, HapMap, 1000 human genome dataset and database of 100 Chinese healthy adults (accessed on February 4, 2022), suggesting the variant may be the pathogenic mutation in this family. The reported mutations in GJA8 were summarized, and the p.E42K mutant was located within the transmembrane domain of the protein (Fig. 3). According to multiple sequence alignments from various species, glutamic acid at position 42 is highly conserved in GJA8 (Fig. 4).

Schematic diagram showing reported human Cx50 mutations (purple bands: missense mutations; blue bands: frame shift (fs) mutations). The red band denotes the mutation p.E42K reported in our study

Multiple sequence alignment of the Cx50 amino acid sequence (codons 26-56) from different species, indicating that glutamic acid at position 42 (red) is highly conserved
Bioinformatics analysis
The homology modeling showed overlapped structure of wild and p.E42K mutant Cx50 gap junction channel from the side view and little change was found on the overall structure of the protein (Fig. 3a). The amino acid conformation was also found to have no evident change (Fig. 3b, c). However, the p.E42K mutation caused negatively charged amino acid become positively charged amino acid, and the surface potential diagram of proteins changed (Fig. 5a, b, c, d and e).

Structure homology modeling of GJA8 a Side view of p.E42K mutant GJA8 in cartoon form, showing little change on the overall structure of the protein. b Local structure of wild-type GJA8. c Local structure of mutant GJA8, had no evident change compared with wild-type (b). d The surface potential diagram of wild-type GJA8, white circle highlights codon 42. e The surface potential diagram of mutant GJA8, changed significantly compared with wild-type (d), the p.E42K mutation caused negatively charged amino acid become positively charged amino acid. Red: negative charge; blue: positive charge
Bioinformatics prediction programs were used to assess the functional effects of the p.E42K mutation in Cx50. EIGEN, MVP, MutPred and REVEL got a result of ‘Pathogenic’, MutationTaster got a result of ‘Disease Causing’, LRT got a result of ‘Deleterious’, and BayesDel addAF, BayesDel noAF, DEOGEN2, FATHMM, MetaLR, MetaSVM, PROVEAN, PrimateAI and SIFT showed a result of ‘Damaging’.
Discussion
In this study, by targeted region sequencing (BGI MGIEasy V4 chip, containing 778 genes associated with ocular diseases through OMIM), we identified a novel heterozygous mutation, NM_005267.5(GJA8):c.124G > A, which is responsible for autosomal dominant congenital cataract in a four-generation Chinese family.
The Gap junction protein alpha 8 (GJA8) gene, encoding 433 amino acid residues of Cx50, is localized on chromosome 1q21.1 (MIM 600,897, NG_016242.1). Just like other connexins, Cx50 is a transmembrane protein that consists of four hydrophobic transmembrane domains (TM1, TM2, TM3, TM4), two conserved extracellular loops (ECL1, ECL2), and three intracellular regions (a cytoplasmic loop, the NH2 and COOH terminal) [11]. Mutations in GJA8 have been known to be associated with congenital cataract in humans. To date, more than 80 GJA8 mutations have been detected in inherited cataract pedigrees (https://cat-map.wustl.edu/). Most of the mutations are located in two extracellular loops (ECL1, ECL2) and COOH terminal while few were reported in transmembrane domains of Cx50 protein.
In our study, the substitution of the positively charged lysine for the negatively charged glutamic acid at position 42 lies on the first transmembrane domain (TM1).The transmembrane domains are responsible for span of plasma membrane and formation of the aqueous pore [12]. Then TM1 is proposed to participate in the oligomerization into connexin hemichannels and the correct transportation of the protein into the plasma membrane [13]. It has been identified that charged pore-lining residues lie in the end of TM1, which are involved in the voltage dependence as a part of the voltage sensor of the slow gate in gap junctions. When glutamate at codon 42 was mutated by a lysine, the profile of the electric field across the hemichannel pore could be changed and the voltage dependence of the hemichannel might be increased [14], thus interfering with the formation of functional gap junctions and leading to cataract formation.
According to the results of bioinformatics prediction, the p.E42K mutation was predicted to have damage effects to the function of GJA8, which emphasized the functional importance of this site. In Xenopus oocytes, Pal et al. [15] found that even one single mutant subunit in a gap junction could inhibit channel function, and several reports revealed GJA8 mutations could inhibit hemichannels [10, 16,17,18]. Connexin hemichannels can protect lens fiber cells against oxidative damage through a unique cell protective mechanism by transporting the extracellular reductant to the intracellular space. When hemichannels are inhibited, the transportation of reductant from extracellular space to intracellular space will be decreased, thus attenuating the protective effect against oxidative stress which will cause lens cells apoptosis and cells death [19, 20]. Collectively, the mutant will inhibit hemichannels activity and reduce cell tolerance to oxidative stress, resulting in protein aggregation, loss of lens cell function and ultimately cataract formation. However, the exact mechanism of our novel GJA8 mutation causing congenital cataract needs further functional analysis to confirm.
Conclusions
In conclusion, our current study is the first to report that NM_005267.5(GJA8):c.124G > A mutation is associated with congenital cataract. Our finding can broaden the mutation spectrum of GJA8, enrich the phenotype-genotype correlation of congenital cataract and help to better understand the genetic background of congenital cataract.
Availability of data and materials
The data that support the findings of this study are not publicly available due to their containing information that could compromise the privacy of patient but are available from the corresponding author (WL) upon reasonable request.
Abbreviations
- GJA8 :
-
Gap junction protein alpha-8
- TM1:
-
The first transmembrane domain
- PolyPhen-2:
-
Polymorphism Phenotyping v2
- PROVEAN:
-
Protein Variation Effect Analyzer
References
Pichi F, Lembo A, Serafino M, Nucci P. Genetics of congenital cataract. Dev Ophthalmol. 2016;57:1–14.
Santana A, Waiswo M. The genetic and molecular basis of congenital cataract. Arq Bras Oftalmol. 2011;74:136–42.
Sheeladevi S, Lawrenson JG, Fielder AR, Suttle CM. Global prevalence of childhood cataract: a systematic review. Eye (Lond). 2016;30:1160–9.
Gilbert C, Foster A. Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ. 2001;79:227–32.
Zhang XH, Da Wang J, Jia HY, Zhang JS, Li Y, Xiong Y, et al. Mutation profiles of congenital cataract genes in 21 northern Chinese families. Mol Vis. 2018;24:471–7.
Li J, Chen X, Yan Y, Yao K. Molecular genetics of congenital cataracts. Exp Eye Res. 2020;191:107872.
Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19:134–49.
Shiels A, Hejtmancik JF. Molecular genetics of cataract. Prog Mol Biol Transl Sci. 2015;134:203–18.
Liu W, Guo R, Hao H, Ji J. Identification of a novel RHO heterozygous nonsense mutation in a Chinese family with autosomal dominant retinitis pigmentosa. BMC Ophthalmol. 2021;21:360.
Li D, Xu C, Huang D, Guo R, Ji J, Liu W. Identification and functional analysis of a novel missense mutation in GJA8, p.Ala69Thr. BMC Ophthalmol. 2020;20:461.
Krysko DV, Leybaert L, Vandenabeele P, D’Herde K. Gap junctions and the propagation of cell survival and cell death signals. Apoptosis. 2005;10:459–69.
Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–400.
Kronengold J, Trexler EB, Bukauskas FF, Bargiello TA, Verselis VK. Single-channel SCAM identifies pore-lining residues in the first extracellular loop and first transmembrane domains of Cx46 hemichannels. J Gen Physiol. 2003;122:389–405.
Pinto BI, Garcia IE, Pupo A, Retamal MA, Martinez AD, Latorre R, et al. Charged residues at the first transmembrane region contribute to the voltage dependence of the slow gate of connexins. J Biol Chem. 2016;291:15740–52.
Pal JD, Berthoud VM, Beyer EC, Mackay D, Shiels A, Ebihara L. Molecular mechanism underlying a Cx50-linked congenital cataract. Am J Physiol. 1999;276:C1443–6.
Beahm DL, Hall JE. Hemichannel and junctional properties of connexin 50. Biophys J. 2002;82:2016–31.
Cui X, Zhou Z, Zhu K, Feng R, Han J, Li M, et al. A Novel Cx50 insert mutation from a Chinese congenital cataract family impairs its cellular membrane localization and function. DNA Cell Biol. 2018;37:449–56.
Shi W, Riquelme MA, Gu S, Jiang JX. Connexin hemichannels mediate glutathione transport and protect lens fiber cells from oxidative stress. J Cell Sci. 2018;131:jcs212506.
Berthoud VM, Beyer EC. Oxidative stress, lens gap junctions, and cataracts. Antioxid Redox Signal. 2009;11:339–53.
Brennan LA, McGreal RS, Kantorow M. Oxidative stress defense and repair systems of the ocular lens. Front Biosci (Elite Ed). 2012;4:141–55.
Acknowledgements
We thank the family members for participating in our study.
Funding
This work was supported by a grant from Open Project of Tianjin Key Laboratory of Retinal Functions and Diseases (2020tjswmq003), Youth Special Fund of Clinical Research of Tianjin Medical University Eye Hospital (2020QN02) and Tianjin Key Medical Discipline (Specialty) Construction Project. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Author information
Authors and Affiliations
Contributions
WL and JJ designed and supervised the study; DH and RG collected the data and drafted the original manuscript; WL, DH and RG analyzed the data and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research followed the tenets of the Declaration of Helsinki, written informed consent was obtained from all the subjects after explanation of the nature and possible consequences of the study. This study was approved by the ethics committee of Tianjin Medical University Eye Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, R., Huang, D., Ji, J. et al. A novel mutation GJA8 NM_005267.5: c.124G > A, p.(E42K) causing congenital nuclear cataract. BMC Ophthalmol 22, 172 (2022). https://doi.org/10.1186/s12886-022-02386-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-022-02386-y