
Abstract
Background
Mucopolysaccharidosis type VI (MPS VI) is a rare autosomal recessive inherited disease caused by mutations in the arylsulfatase B (ARSB) gene. MPS VI is a multisystemic disease resulting from a deficiency in arylsulfatase B causing an accumulation of glycosaminoglycans in the tissues and organs of the body.
In this report, we present the case of a 16-year-old Chinese male who presented with vision loss caused by corneal opacity. MPS VI was confirmed by genetic diagnosis.
Case presentation
A 16-year-old Chinese male presented with a one-year history of binocular vision loss. The best-corrected visual acuity was 0.25 in the right eye and 0.5 in the left eye. Although slit-lamp examination revealed corneal opacification in both eyes, the ocular examinations of his parents were normal. At the same time, the patient presented with kyphotic deformity, short stature, joint and skeletal malformation, thick lips, long fingers, and coarse facial features. Genetic assessments revealed that ARSB was the causative gene. Compound heterozygous missense mutations were found in the ARSB gene, namely c.1325G > A (p. Thr442Met) (M1) and c.1197G > C (p. Phe399Leu) (M2). Genetic diagnosis confirmed that the patient had MPS VI.
Conclusions
This paper reports a case of MPS VI confirmed by genetic diagnosis.
MPS VI is a multisystem metabolic disease, with corneal opacity as a concomitant ocular symptom. As it is difficult for ophthalmologists to definitively diagnose MPS VI, genetic testing is useful for disease confirmation.
Similar content being viewed by others

Background
Mucopolysaccharidosis type VI (MPS VI) is a rare lysosomal storage disease caused by deficient activity of arylsulfatase B (ARSB) [1]. The ARSB gene encodes the lysosomal enzyme ARSB, also known as N-acetylgalactosamine 4-sulfatase. This gene (NM_000046.5) is located on chromosome 5q13-q14 and contains eight exons and seven introns encoding 533 amino acids [2]. The abnormal accumulation of glycosaminoglycans (GAGs), dermatan sulfate, and chondroitin 4-sulfate resulting from a deficiency of ARSB could lead to MPS VI, which involves multiple organ systems and displays genetic and phenotypic heterogeneity [3]. This disease has various clinical manifestations, including short stature, corneal opacity, cardiac abnormalities, coarse facial features, hepatosplenomegaly, multifunctional disorders, joint stiffness, spinal protrusion, thick lips, and long fingers [4]. Here, we report the case of a 16-year-old patient who presented with vision loss; clinical examination revealed that the patient had corneal opacity and physical deformities. MPS VI was ultimately diagnosed by genetic analysis.
Case presentation
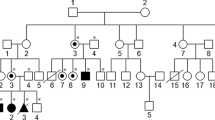
A 16-year-old male patient visited our ophthalmology department due to deteriorating vision in both eyes over the past year. His parents were not related to one another, he was delivered by cesarean section after 38 weeks and weighed 2600 g at birth, and his parents and sister presented with normal vision and stature (Fig. 1a). His parents said the patient was normal at birth, but developed skeletal abnormalities at age one. When he was 9 years old, the patient visited the Beijing Children’s Hospital, but there was no definitive diagnosis. Slit-lamp examination of both eyes demonstrated corneal haze (Figs. 1b and c), and a Goldmann applanation tonometer showed that his intraocular pressure was 26.7 mmHg and 17.3 mmHg in his right and left eye, respectively. Uncorrected visual acuity was 0.25 in his right eye and 0.5 in his left eye. Of note, his best-corrected visual acuity did not increase: right eye + 4.5 DS/− 1.5 DC = 0.25, left eye + 3.5 DS/− 2.0 DC = 0.5. The axial length measured using an ultrasonic pachymeter showed the right eye to be 20.39 mm and the left eye to be 20.27 mm. Endothelial cell counts could not be determined due to the corneal opacity of the patient. Although the patient exhibited normal intelligence, physical examination revealed kyphotic deformities, short stature (compared to peers), skeletal and joint deformity, sternum herniation, thick lips, long fingers, coarse facial features, and a flat nasal bridge. Taken together, we could not provide a definite diagnosis according to the ocular manifestations with systemic manifestations, but we suspected that he had a rare hereditary syndrome. Therefore, we recommend the consanguineous family to make genetic diagnosis. All subjects provided signed informed consent, this study was approved by the Ethics Committee of the Affiliated Hospital of Yunnan University and was performed in accordance with the Declaration of Helsinki. Peripheral venous blood samples (5 ml) were drawn from the patient and his parents for genetic testing.

Pedigree and clinical examination of the proband. a Males are denoted with squares and females with circles. Empty and filled symbols indicate unaffected and affected individuals, respectively. Arrow represents the proband. b, c Anterior segment photographs of the (b) right and (c) left eye of the proband show corneal opacification in both eyes
Genetic assessments revealed two heterozygous missense mutations in the ARSB gene, exon 7 c.1325G > A (p.Thr442Met) (M1) and exon 6 c.1197G > C (p.Phe399Leu) (M2), co-segregated with the disease phenotype in this family, and his healthy parents were heterozygous carriers (father: c.1197G > C, mother: c.1325G > A) (Figs. 2a and b).

Mutation analysis of ARSB. a, b There were two mutations in the ARSB gene, i.e. c.1325G > A (p.Thr442Met) (M1) and c.1197G > C (p.Phe399Leu) (M2). Sequencing results showed that c.1325G > A (p. Thr442Met) was inherited from the mother, whereas c.1197G > C (p. Phe399Leu) was inherited from the father. c, d Mutant amino acids (No. 399, phenylalanine and No. 442, threonine) are highly conserved across species. e, f, g, h Molecular modeling of the (e, f) wild-type and (g, h) mutant ARSB protein. Structural modeling of the ARSB protein demonstrated that mutation of amino acid No. 399 (phenylalanine) resulted in the breakage of hydrogen bonds, and mutation of amino acid No. 442 (threonine) resulted in the partial breakage of hydrogen bonds
The mutated areas were found in a highly conserved segment of the ARSB protein in humans and other species, and affected amino acids 399 (phenylalanine) and 442 (threonine) (Figs. 2c and d). According to the American College of Medical Genetics and Genomics, and analysis of four bioinformatics platforms (SIFT, Mutation Taster, Polyphen2, and REVEL), M1 was proposed as a pathogenic mutation and M2 was classified as likely pathogenic [5]. The global MAF of p.Phe399Leu was low [MAF(gnomAD) = 0.003]; however, the global MAF of p.Thr442Met was not found in the databases. Structural modeling of the ARSB protein (Figs. 2e and f) demonstrated that both Phe399 and Thr442 were located in the first β-sheet, which was required for receptor binding. The mutation caused the hydrogen bond between the amino acids to break (Figs. 2g and h), which resulted in protein structural instability. The patient was confirmed to have MPS VI because of the compound heterozygous mutations of ARSB.
Discussion
Approximately 163 mutations of ARSB have been reported to date, among which missense and nonsense mutations are the most common [6]. In concordance with the mutation of ARSB and the lack of ARSB activity, GAGs cannot be completely degraded in lysosomes, which leads to the deposition of its substrates, further resulting in MPS VI [7]. Patients with MPS VI show light or heavy development with a varying age of onset and disease progression. The early diagnosis of patients with lightly-developed MPS is challenging, as they have a slow progression and no typical clinical features of MPS VI in the early stages of the disease [8]. Heavy MPS VI typically has an age of onset before 2 or 3 years, and patients usually present with short stature, scoliosis, cardiopulmonary dysfunction, cervical spinal cord compression, coarse facial features, and normal intelligence; these patients typically die from cardiopulmonary complications between the ages of 20 and 30 years [9]. Swiedler et al. presented a multicenter and multinational study of up to 123 patients, and suggested that the rate of disease progression in MPS VI could be determined based on urinary glycosaminoglycan (uGAG) levels. When the uGAG to creatinine ratio was > 100 μg/mg, such patients exhibited rapid disease progression and died before the age of 20 years. If the uGAG to creatinine ratio was < 100 μg/mg, these patients had a longer survival time [10]. MPS VI diagnosis can be confirmed by detecting ARSB activity in peripheral blood leukocytes or fibroblasts, or through ARSB gene testing, which not only facilitates further diagnosis, but also helps to elucidate the relationship between genotype and phenotype [6]. Compound heterozygous mutations exon 7 c.1325G > A (p.Thr442Met) and exon 6 c.1197G > C (p.Phe399Leu) in ARSB causing MPS VI have been previously reported, with Chupong et al. presenting data on four MPS VI patients, one of patient exhibited novel compound heterozygous missense ARSB mutations [11]. In the present case, we report the case of a 16-year-old male patient with MPS VI, which was confirmed by genetic testing. Both patient and his parents were genetically tested in this study, and two mutations were identified in the ARSB gene, namely c.1325G > A (p.Thr442Met) (M1) and c.1197G > C (p.Phe399Leu) (M2). The mutations inherited by the proband from his healthy parents were confirmed by family separation verification. Corneal opacity is a common clinical symptom of MPS VI that can present early on in the disease; such patients can develop other ocular complications, including glaucoma and optic neuropathy [12]. Thus, clinical ophthalmologists should possess increased disease awareness. Moreover, ophthalmologists should adequately assess the general condition of patients and note the family history of hereditary diseases when assessing corneal turbidity in patients who display concomitant growth and development disorders and skeletal abnormalities. If necessary, genetic testing should be undertaken to avoid misdiagnosis and missed diagnosis [13]. Overall, the present report details the confirmed diagnosis of MPS VI through genetic testing in a patient who presented with vision loss.
Availability of data and materials
All data generated and/or analysed during this study are included in this published article and sanger sequencing results are in additional supporting files.
Abbreviations
- MPS VI:
-
Mucopolysaccharidosis type VI
- ARSB:
-
Arylsulfatase B
References
Lin WD, Lin SP, Wang CH, Hwu WL, Chuang CK, Lin SJ, et al. Genetic analysis of mucopolysaccharidosis type VI in Taiwanese patients. Clin Chim Acta. 2008;394(1–2):89–93.
Modaressi S, Rupp K, von Figura K, Peters C. Structure of the human arylsulfatase B gene. Biol Chem Hoppe Seyler. 1993;374(5):327–35.
Harmatz P, Shediac R. Mucopolysaccharidosis VI: pathophysiology, diagnosis and treatment. Front Biosci (Landmark edition). 2017;22:385–406.
Hancer VS, Buyukdogan M, Babameto-Laku A. A novel pathological ARSB mutation (c.870G>a; p.Trp290stop) in Mucopolysaccharidosis type VI patients. Mol Syndromol. 2020;10(5):272–5.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Zapała B, Chmura O, Ciałowicz U, Solnica B, Krajewska-Włodarczyk M, Żuber Z. A case of mucopolysaccharidosis type VI in a polish family. Importance of genetic testing and genotype-phenotype relationship in the diagnosis of mucopolysaccharidosis. Mol Genet Metab Rep. 2020;25:100658.
CADTH Common Drug Reviews. In: Galsulfase (Naglazyme). edn. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health Copyright © CADTH 2016.; 2016.
Thümler A, Miebach E, Lampe C, Pitz S, Kamin W, Kampmann C, et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis. 2012;35(6):1071–9.
Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5.
Swiedler SJ, Beck M, Bajbouj M, Giugliani R, Schwartz I, Harmatz P, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). Am J Med Genet Part A. 2005;134a(2):144–50.
Ittiwut C, Boonbuamas S, Srichomthong C, Ittiwut R, Suphapeetiporn K, Shotelersuk V. Novel mutations, including a large deletion in the ARSB gene, causing Mucopolysaccharidosis type VI. Genet Test Mol Biomark. 2017;21(1):58–62.
Koseoglu ST, Harmatz P, Turbeville S, Nicely H. Reversed papilledema in an MPS VI patient with galsulfase (Naglazyme) therapy. Int Ophthalmol. 2009;29(4):267–9.
Summers CG, Ashworth JL. Ocular manifestations as key features for diagnosing mucopolysaccharidoses. Rheumatology (Oxford). 2011;50(Suppl 5):v34–40.
Acknowledgments
We appreciate all patients and family members for their participation in this study.
Funding
This study was supported by the Yunnan provincial Major Science and Technology Foundation [Grant No. 2018ZF009] and the Eye Disease Clinical Medical Center of Yunnan province [Grant No. YXZX-2019058]. This funding was used to support data collection and interpretation, as well as writing of this manuscript.
Author information
Authors and Affiliations
Contributions
HL and MFH designed and conducted the study. YTW provided clinical care. JY and MJD analyzed and interpreted the patient data. MFH, JY, and HL were major contributors to writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patient’s legal guardian provided written, informed consent prior to undergoing this study. This study was approved by the ethics committee of The Second People’s Hospital of Yunnan Province, Affiliated Hospital of Yunnan University (No.2020089).
Consent for publication
The risk of identification of the patient was minimized and the patient’s legal guardian provided written, informed consent for publication of this report.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, MF., Yang, J., Dong, MJ. et al. Compound heterozygous missense mutations in a Chinese mucopolysaccharidosis type VI patient: a case report. BMC Ophthalmol 21, 214 (2021). https://doi.org/10.1186/s12886-021-01979-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-021-01979-3