
Abstract
Background
Retinitis pigmentosa (RP) is the most common inherited retinal dystrophy, affecting approximately 1 in 4000 individuals worldwide. The most common form of syndromic RP is Usher syndrome (USH) accounting for approximately 20–30 % of RP cases. Mutations in the USH2A gene cause a significant proportion of recessive non-syndromic RP and USH type II (USH2). This study aimed to determine the causative role of the USH2A gene in autosomal recessive inherited ocular diseases and to establish genotype-phenotype correlation associated with USH2A variants.
Methods
We performed direct Sanger sequencing and co-segregation analysis of the USH2A gene to identify disease causing variants in a non-syndromic RP family, two USH2 families and two Keratoconus (KC) families.
Results
Disease causing variants in the USH2A gene were identified in two families displayed KC and USH2 phenotypes. A novel variant c.4029T > G, p.Asn1343Lys in the USH2A gene was detected in a Pakistani family with KC phenotype. In addition, a missense variant (c.7334 C > T, p. Ser2445Phe) in the USH2A gene was found segregating in another Pakistani family with USH2 phenotype. Homozygosity of identified missense USH2A variants was found associated with autosomal recessive inherited KC and USH2 phenotypes in investigated families. These variants were not detected in ethnically matched healthy controls. Moreover, the USH2A variants were predicted to be deleterious or potentially disease causing by PolyPhen-2, PROVEAN and SIFT.
Conclusions
This study provided first evidence for association of a novel USH2A variant with KC phenotype in a Pakistani family as well as established the phenotype-genotype correlation of a USH2A variant (c.7334 C > T, p. Ser2445Phe) with USH2 phenotype in another Pakistani family. The phenotype-genotype correlations established in present study may improve clinical diagnosis of affected individuals for better management and counseling.
Similar content being viewed by others


Background
Inherited retinal dystrophies (IRDs) are a broad group of clinically and genetically heterogeneous rare eye diseases resulting in progressive visual impairment or blindness [1]. Retinitis pigmentosa (RP; MIM#268,000) is the most prevalent form of IRDs, affecting approximately 1 in 4000 individuals worldwide [2]. RP is characterized by progressive bilateral degeneration of the rod and cone photoreceptors that leads to night blindness and progressive visual field defects [3]. Majority of RP cases are non-syndromic, whereas in 20–30 % cases are syndromic [4]. The most common form of syndromic RP is Usher syndrome (USH) and the general prevalence of USH approximately ranges from 1 to 4 in 25,000 individuals [5]. USH is characterized by congenital severe-to-profound sensorineural hearing loss with RP and rarely vestibular dysfunction. Depending on the severity, progression and age of onset of these features USH is divided into three clinical types: USH type 1 (USH1:MIM #276,901), USH type 2 (USH2: MIM#276,902) and USH type 3 (USH3:MIM#276,903), [6]. Each USH subtype is also genetically heterogeneous and several genes have been described for all three types.
The USH2A gene is located on chromosome 1q41 and has two alternatively spliced isoforms: a short isoform A and a long isoform B. A 5 kb short isoform is transcribed from 1to 21 exons of USH2A and translated into a putative 170 kDa extracellular secreted protein of 1,551 amino acids [7] that is expressed in both the retina and the inner ear [8]. A 15 kb long isoform is transcribed including additional 51 exons at the 3′ end of USH2A and translated into a 600-kDa transmembrane protein of 5202 amino acids [9, 10]. The dominant form of the USH2A protein, also known as usherin in photoreceptors is the 600-kDa polypeptide [9] that shows homology to both extracellular matrix proteins and receptors containing 10 laminin EGF-like domains, 35 fibronectine type-III (FN3) motifs, two laminin G domains and a PDZ-binding motif [9, 11]. This protein is expressed in adult human retina, specially localized to the photoreceptor cells and is required for the long-term maintenance of retinal photoreceptors and for the development of fetal cochlear hair cells [12]. The USH2A variants have been reported to cause recessive RP in 23 % cases [13], USH2 in 85 % cases [14]. In addition, the recessive USH2A variants have also been reported to cause non-syndromic hearing loss or deafness in few cases [15, 16]. According to our knowledge, no specific study has reported the association of USH2A variants with corneal diseases like Keratoconus (KC).However, studies have provided evidence for integration of USH2A into a protein network that is important in development and maintenance of the inner ear and retina [17–20]. In addition, mutations within USH2A affect its interaction with other proteins in network and may cause a broad spectrum of phenotypes in the inner ear and eye [18]. Therefore, to gain more insights into USH2A associated ocular diseases, we screened USH2A in Pakistani families with RP, USH2 and KC phenotypes in this study.
We identified a homozygous missense variants c.4029T > G, p.Asn1343Lys and c.7334 C > T, p. Ser2445Phe in the USH2A gene that segregated with KC and USH2 phenotypes in Pakistani families.
Materials and methods
Ethical approval and consent to participate
Approval was obtained from the Ethical Committee of Kohat University of Science and Technology (KUST), and the study was carried out in accordance with the Declaration of Helsinki.Informed written consent was obtained for participation in the study from families’ members and parents of the minor children. Families contained five living individuals and at least two living individuals with any type of rare inherited retinal or corneal disease with onset before age 20 years were eligible for the study. Following eligibility criteria, one non-syndromic RP, two USH2 and two KC families were recruited with help of ophthalmologists from the Khyber Pakhtunkhwa region of Pakistan. Demographic characteristics of recruited families are shown in Table 1.The phenotypically affected individuals in participated families underwent thorough ophthalmologic examinations for confirmation of clinical diagnosis of non-syndromic RP, USH and KC in them. The participated families were visited at the place of residence, pedigrees were drawn and disease associated features were recorded. Blood samples were collected from affected and normal individuals of both families. Blood samples were also collected from 100 ethnically matched unrelated healthy Pakistani individuals and were used as controls for allele frequencies and confirmation of disease associated variants.
Detection of USH2A sequence variants
Genomic DNA from the blood samples was extracted using the GenElute™ Blood Genomic DNA kit (Sigma-Aldrich.com) according to the manufacturer’s protocol.
In this study direct Sanger sequencing and co-segregation analysis was performed to identify disease causing variants in the USH2A gene in all the recruited families. The coding region comprising of 2 to 72 exons and their intron–exon boundaries of the USH2A gene were amplified by polymerase chain reaction (PCR) in these families using primers reported previously [21], following standard conditions. The PCR amplified products were sequenced by the Macrogen sequencing service (Seoul, South Korea) in order to check the co-segregation of the variants with the disease phenotype in investigated families. The potential pathogenicity of each segregating sequence variant was determined using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), PROVEAN (http://provean.jcvi.org/index.php) and SIFT (http://sift.jcvi.org/) specialized prediction software. The conservation of a particular amino acid at a specific position was determined using Ensemble Wasabi viewer software (https://www.ensembl.org/index.html). To compare and correlate each USH2A gene variant with the disease phenotype, all reported variants were retrieved from HGMD (http://www.hgmd.cf.ac.uk/ac/search.php), OMIM (https://www.ncbi.nlm.nih.gov/omim/) and PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) databases.
Results
Clinical characteristics
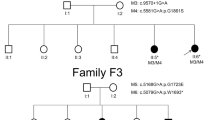
Disease causing variants in the USH2A gene were identified in a four-generation KC family and a two-generation USH2 family as shown in Fig. 1 (i) and (ii) a.Ophthalmology reports on affected members of both families confirmed clinical diagnosis of KC and USH2. The four affected individuals I: 1, III: 1, III: 5 and III: 8 were of 75, 37, 25 and 20 years of age respectively at the time of recruitment of KC family. KC occurred around 15 years of age in four affected individuals with symptoms include mild astigmatism, myopia, photophobia and eye itching and the degree of vision impairment increased with their increasing age. However, individuals younger than 10 years of age (IV: 1, IV: 2 and IV: 3) had no signs and symptoms of KC. The affected individuals in KC family were fitted with spectacle correction and two of them (III: 1 and III: 5) underwent the corneal cross linking surgery before age 21 years.

Family pedigrees, genotypes, and USH2A variants. (i) a. KC Family and (ii) a. USH2 family. In pedigrees, squares symbolize males and circles symbolize females. All filled circles and squares symbolize affected members, whereas clear circles and squares symbolize unaffected members. In addition, pedigrees affected by USH2A variants showing segregation of the altered alleles. (i) and (ii) b. Sequence chromatograms showing wild type and USH2A [c.4029T > G, p.Asn1343Lys (KC family), and, c.7334 C > T, p. Ser2445Phe (USH2 family)] variants. (i) and (ii) c. Multiple alignments of the partial amino acid sequences of USH2A in a variety of vertebrate and non-vertebrate species, show stringent conservation of Asparagine at position 1343 and Serine at position 2445
The three affected individuals II: 3, III: 1 and III: 2 were of 57, 29 and 27 years of age respectively at the time of recruitment of USH2 family. The affected individuals had bilateral, congenital, severe sensorineural hearing loss, normal vestibular function and symptoms of progressive RP. The affected individuals with USH2 experienced night blindness and progressive bilateral visual loss in early adolescence. Affected individuals (III: 1 and III: 2) were fitted with bilateral hearing aids and their affected mother (II:3) was blind and deaf at the time of recruitment of USH2 family.
USH2A sequence variants
Mutation screening of the USH2A gene in KC family revealed a novel missense homozygous variant (NM_206933.4:c.4029T > G) in exon 17 condition in four affected individuals as well as in a phenotypically normal 6 years old child as shown in Fig. 1 (i) a and b. The variant c.4029T > G leads to a substitution of asparagine with lysine at the evolutionary conserved position 1343 (NP_996816.3:p.Asn1343Lys) according to the Ensemble databases as shown in Fig. 1(i) c. Insilco analysis of p.Asn1343Lys using PolyPhene-2, PROVEAN and SIFT predicted it as probably damaging or deleterious (Table 2).
Direct sequencing analysis of the USH2A gene in USH2 family identified a missense homozygous variant (NM_206933.4: c.7334 C > T) in exon 19 in affected individuals and unaffected father and siblings were detected carriers for this variant as shown in Fig. 1 (ii) a and b. This single base pair change is predicted to result in substitution of serine with phenylalanine at evolutionary conserved residue position 2445 (p.Ser2445Phe) as shown in Fig. 1c (ii). This p.Asn1343Lys variant was also predicted probably damaging or deleterious or potentially disease causing by PolyPhene-2, PROVEAN and SIFT respectively (Table 2).
The identified USH2A variants in KC and USH2 families were found segregating in an expected autosomal recessive manner as shown in Fig. 1 (i) and (ii) a and were not detected in Pakistani unrelated healthy controls. A schematic representation of USH2A encoded usherin protein is presented to show spectrum of sequence variants identified in present study and previously reported in Pakistani families for various inherited ocular diseases in Fig. 2.

Schematic representation of domains of predicted protein product, highlighting the positions of all disease associated USH2A variants identified in Pakistani families to date. Discrete color pattern of variants shows type of phenotype
Discussion
USH2A variants have been described to cause most commonly syndromic and non-syndromic forms of recessive IRD in different populations of world. High frequencies of USH2A variants have been reported in USH2 families of Jewish, Spanish, American, Scandinavian, Slovenian, and Italian, British origins [22–28]. In addition, USH2A variants have been identified to cause a substantial number of non-syndromic RP in families of Caucasian, Japanese, American and Chinese origins [13, 25, 29–33]. Importantly, USH2A variants display a wide phenotypic spectrum, therefore, phenotype-genotype correlation for the most prevalent USH2A variants may facilitate genetic counselling and improve the prognosis of affected individuals, as well as guide for patient-specific treatment options [26].
Available evidences suggest presence of USH2A recurrent variants in different populations as a result of founder effect. Of the 896 previously identified and previously reported USH2A variants in the Human Gene Mutation Database (HGMD), the p.Cys759Phe have been described the most prevalent pathogenic variant in the Spanish population and accounts for 4.5 % of the RP [34, 35] and 8.1 % of the USH2 cases [36], Similarly, a USH2A variant p.Trp3955Terwas detected in majority of Slovenian USH2 cases [26]. A deleterious USH2A variant p.Glu767fs have been found associated most commonly with USH2 [24, 37] and significantly with non-syndromic RP Caucasian patients [38]. A splice site founder variant (c.8559-2 A > G) of the USH2A was reported in majority of Chinese and Japanese USH2 patients [39, 40]. In addition, four USH2A variants c.239-240insGTAC, c.1000 C > T, c.2209 C > T, and c.12067-2 A > G account for 64 % USH2 in Jewish families of non-Ashkenazi descent [22]. A founder effect has been identified for a USH2A variant p.Cys1447fs that accounts for 55.6 % of the USH2 cases among Quebec French-Canadians [41]. However, great majority of reported USH2A variants are rare private mutations segregating in single families from different ethnic populations worldwide [42–45]. Involvement of USH2A private variants in causing profound hearing loss or deafness and IRDs in Pakistani families has been described in previous studies [16, 46,47,48]
Here, we report a novel missense private variant (c.4029T > G, p.Asn1343Lys) in the USH2A gene in a Pakistani family with KC phenotype. This variant was not found in population databases and was predicted deleterious or probably disease causing by in silico tools. The co-segregational analysis revealed that the four affected individuals and a clinically asymptomatic 6 years old individual in KC family were found homozygous for the identified variant. Importantly, this phenotype genotype difference in an individual could be attributed to the fact that the age of onset of disease symptoms was observed 15 years in investigated family. In this study we also report a missense variant (c.7334 C > T, p. Ser2445Phe) in the USH2A gene in another Pakistani family with USH2 phenotype. Noteworthy, the c.7334 C > T, p. Ser2445Phe variant was previously identified by Carss et al. [49] in a compound heterozygous form in a single individual with RP and present study provides evidence of this variant correlation with USH2 phenotype. It is a fact that both USH2 and non-syndromic RP are the most prevalent phenotypic variants of IRD caused by autosomal recessive USH2A variants, the findings of current investigation and previous one by Carss et al. [49] clearly demonstrate this fact and additionally, excluded the presence of a putative genetic modifier factor within the USH2A gene that may contribute in the development of two phenotypes.
Till date, mutation screening in Pakistani families revealed in total seven USH2A variants by previous researches [16, 46,47,48]. The USH2A variants described previously and in present study in Pakistani families thereby affecting functional residues absolutely conserved in the different domains of usherin protein. Among the two homozygous pathogenic USH2A variants identified in present study, a KC associated variant p.Asn1343Lys is located in the FN3 motif 2 and a USH2 associated variant p.Ser2445Phe is located in the FN3 motif 11. Previously reported homozygous pathogenic variants p.Gln1221Ter, p.Arg1914Cys, p.His3825fs, and p.Trp4175Gly in Pakistani families are located in the FN3 motifs 2,5,23 and 27 respectively. A compound heterozygous (p.Pro2811Thr and p. Pro3504Ala) variant identified in the USH2A gene in another Pakistani family was predicted to affect the FN3 motifs 14 and 20 respectively. All these variants reported in this study and previously affect highly conserved residues in the FN3 motifs across model organisms including human. The FN3 motifs have been shown to be involved in binding substrates thus highlighting the functional importance of the FN3 motifs in substrate selectivity. In addition, a nonsense USH2A variant p.Arg1549Ter identified previously in a Pakistani family [47], is located in the Laminin G-like 1 domain that result in production of truncated protein product and severe reduction in abundance of cellular transcript due to targeted degradation by nonsense-mediated decay.
Establishing the diagnosis for ocular diseases with greater clinical and genetic heterogeneity is quite difficult and challenging. This problem is usually more common in developing countries such as Pakistan, where people live in rural areas and generally have less access to healthcare and ophthalmic services. There is also limited availability of specific and expensive tests that are required for diagnostic investigations of ocular diseases. Mutation screening in USH2A in the current study enabled an accurate molecular diagnosis of USH2A-associated KC and USH2 to be established and has facilitated informed genetic counselling.
Conclusions
This study revealed a novel genotype–phenotype correlation associated with USH2A (c.4029T > G, p.Asn1343Lys) variant as well as provided sufficient evidence to establish genotype–phenotype correlation of a USH2A variant (c.7334 C > T, p. Ser2445Phe) with USH2 phenotype. Analysis of genotype-phenotype correlation may increase our understanding of the diseases and may help in better management and counseling of affected individuals with USH2A variants and provides new targets for therapeutic approaches.
Availability of data and materials
All the data used to support the findings of this study are included within the article and are available on request from corresponding author.
References
Ginter PM, Duncan WJ, Swayne LE. The strategic management of health care organizations. Hoboken: Wiley; 2018.
den Hollander AI, Black A, Bennett J, Cremers FP. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest. 2010;120(9):3042–53.
Rivolta C, Sharon D, DeAngelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002;11(10):1219–27.
Ferrari F: Health and religious rituals in South Asia: disease, possession and healing: Taylor & Francis; 2011.
Mathur P, Yang J. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. 2015;1852(3):406–20.
Besnard T, García-García G, Baux D, Vaché C, Faugère V, Larrieu L, Léonard S, Millan JM, Malcolm S, Claustres M. Experience of targeted Usher exome sequencing as a clinical test. Mol Genet Gen Med. 2014;2(1):30–43.
Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280(5370):1753–7.
Pearsall N, Bhattacharya G, Wisecarver J, Adams J, Cosgrove D, Kimberling W. Usherin expression is highly conserved in mouse and human tissues. Hearing Res. 2002;174(1–2):55–63.
Van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, Cremers FP, Cremers CW, Kremer H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004;74(4):738–44.
Reiners J, Marker T, Jurgens K, Reidel B, Wolfrum U: Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C). 2005.
Weston M, Eudy JD, Fujita S, Yao S-F, Usami S, Cremers C, Greenburg J, Ramesar R, Martini A, Moller C. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000;66(4):1199–210.
Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, Li T: Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proceedings of the National Academy of Sciences 2007; 104(11):4413–4418.
McGee TL, Seyedahmadi BJ, Sweeney MO, Dryja TP, Berson EL. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J Med Genet. 2010;47(7):499–506.
Yan D, Liu XZ. Genetics and pathological mechanisms of Usher syndrome. J Hum Genet. 2010;55(6):327–35.
Mutai H, Suzuki N, Shimizu A, Torii C, Namba K, Morimoto N, Kudoh J, Kaga K, Kosaki K, Matsunaga T. Diverse spectrum of rare deafness genes underlies early-childhood hearing loss in Japanese patients: a cross-sectional, multi-center next-generation sequencing study. Orphan J Rare Dis. 2013;8(1):172.
Wang R, Han S, Khan A, Zhang X. Molecular analysis of twelve Pakistani families with nonsyndromic or syndromic hearing loss. Genet Test Mol Bio. 2017;21(5):316–21.
Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14(3):347–56.
Kremer H, van Wijk E, Märker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15(suppl_2):R262–70.
Maerker T, van Wijk E, Overlack N, Kersten FF, McGee J, Goldmann T, Sehn E, Roepman R, Walsh EJ, Kremer H. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet. 2008;17(1):71–86.
Tian G, Zhou Y, Hajkova D, Miyagi M, Dinculescu A, Hauswirth WW, Palczewski K, Geng R, Alagramam KN, Isosomppi J. Clarin-1, encoded by the Usher Syndrome III causative gene, forms a membranous microdomain: possible role of clarin-1 in organizing the actin cytoskeleton. J Bio Chem. 2009;284(28):18980–93.
Aller E, Jaijo T, Beneyto M, Najera C, Oltra S, Ayuso C, Baiget M, Carballo M, Antinolo G, Valverde D. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006;43(11):e55–e55.
Auslender N, Bandah D, Rizel L, Behar DM, Shohat M, Banin E, Allon-Shalev S, Sharony R, Sharon D, Ben-Yosef T. Four USH2A founder mutations underlie the majority of Usher syndrome type 2 cases among non-Ashkenazi Jews. Genet Test. 2008;12(2):289–94.
Garcia-Garcia G, Aparisi MJ, Jaijo T, Rodrigo R, Leon AM, Avila-Fernandez A, Blanco-Kelly F, Bernal S, Navarro R, Diaz-Llopis M. Mutational screening of the USH2A gene in Spanish USH patients reveals 23 novel pathogenic mutations. Orphan J Rare Dis. 2011;6(1):65.
Yan D, Ouyang X, Patterson DM, Du LL, Jacobson SG, Liu X-Z. Mutation analysis in the long isoform of USH2A in American patients with Usher Syndrome type II. J Hum Genet. 2009;54(12):732–8.
Dreyer B, Brox V, Tranebjærg L, Rosenberg T, Sadeghi AM, Möller C, Nilssen O. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat. 2008;29(3):451.
Zupan A, Fakin A, Battelino S, Jarc-Vidmar M, Hawlina M, Bonnet C, Petit C, Glavač D. Clinical and Haplotypic Variability of Slovenian USH2A Patients Homozygous for the c. 11864G > A Nonsense Mutation. Genes. 2019;10(12):1015.
Sodi A, Mariottini A, Passerini I, Murro V, Tachyla I, Bianchi B, Menchini U, Torricelli F. MYO7A and USH2A gene sequence variants in Italian patients with Usher syndrome. Mol Vis. 2014;20:1717.
Stabej PLQ, Saihan Z, Rangesh N, Steele-Stallard HB, Ambrose J, Coffey A, Emmerson J, Haralambous E, Hughes Y, Steel KP. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet. 2012;49(1):27–36.
Aller E, Larrieu L, Jaijo T, Baux D, Espinós C, González-Candelas F, Nájera C, Palau F, Claustres M, Roux A-F. The USH2A c. 2299delG mutation: dating its common origin in a Southern European population. Eur J Hum Genet. 2010;18(7):788–93.
Ávila-Fernández A, Cantalapiedra D, Aller E, Vallespín E, Aguirre-Lambán J, Blanco-Kelly F, Corton M, Riveiro-Álvarez R, Allikmets R, Trujillo-Tiebas MJ. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol Vis. 2010;16:2550.
Zhao Y, Hosono K, Suto K, Ishigami C, Arai Y, Hikoya A, Hirami Y, Ohtsubo M, Ueno S, Terasaki H. The first USH2A mutation analysis of Japanese autosomal recessive retinitis pigmentosa patients: a totally different mutation profile with the lack of frequent mutations found in Caucasian patients. J Hum Genet. 2014;59(9):521–8.
Huang L, Mao Y, Yang J, Li Y, Li Y, Yang Z. Mutation screening of the USH2A gene in retinitis pigmentosa and USHER patients in a Han Chinese population. Eye. 2018;32(10):1608–14.
Karali M, Testa F, Brunetti-Pierri R, Di Iorio V, Pizzo M, Melillo P, Barillari MR, Torella A, Musacchia F, D’Angelo L. Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa. Int J Mol Sci. 2020;21(1):86.
Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66(6):1975–8.
Bernal S, Ayuso C, Antinolo G, Gimenez A, Borrego S, Trujillo M, Marcos I, Calaf M, Del Rio E, Baiget M. Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet. 2003;40(1):e8–e8.
Blanco-Kelly F, Jaijo T, Aller E, Avila-Fernandez A, López-Molina MI, Giménez A, García-Sandoval B, Millán JM, Ayuso C. Clinical aspects of Usher syndrome and the USH2A gene in a cohort of 433 patients. JAMA Ophthalmol. 2015;133(2):157–64.
Leroy BP, Aragon-Martin JA, Weston MD, Bessant DA, Willis C, Webster AR, Bird AC, Kimberling WJ, Payne AM, Bhattacharya SS. Spectrum of mutations in USH2A in British patients with Usher syndrome type II. Exp Eye Res. 2001;72:503–9.
González-del Pozo M, Martín-Sánchez M, Bravo-Gil N, Méndez-Vidal C, Chimenea Á, Rodríguez-de la Rúa E, Borrego S, Antiñolo G. Searching the second hit in patients with inherited retinal dystrophies and monoallelic variants in ABCA4, USH2A and CEP290 by whole-gene targeted sequencing. Sci Rep. 2018;8(1):1–12.
Jiang L, Liang X, Li Y, Wang J, Zaneveld JE, Wang H, Xu S, Wang K, Wang B, Chen R. Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphan J Rare Dis. 2015;10(1):110.
Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Mizuta K, Mineta H, Minoshima S. Identification of 11 novel mutations in USH2A among Japanese patients with Usher syndrome type 2. Clin Genet. 2009;76(4):383–91.
Ebermann I, Koenekoop RK, Lopez I, Bou-Khzam L, Pigeon R, Bolz HJ. An USH2A founder mutation is the major cause of Usher syndrome type 2 in Canadians of French origin and confirms common roots of Quebecois and Acadians. Eur J Hum Genet. 2009;17(1):80–4.
Kaiserman N, Obolensky A, Banin E, Sharon D. Novel USH2A mutations in Israeli patients with retinitis pigmentosa and Usher syndrome type 2. Arch Ophthalmol. 2007;125(2):219–24.
Perez-Carro R, Corton M, Sánchez-Navarro I, Zurita O, Sanchez-Bolivar N, Sánchez-Alcudia R, Lelieveld SH, Aller E, Lopez-Martinez MA, López-Molina MI. Panel-based NGS reveals novel pathogenic mutations in autosomal recessive retinitis pigmentosa. Sci Rep. 2016;6:19531.
He C, Liu X, Zhong Z, Chen J. Mutation screening of the USH2A gene reveals two novel pathogenic variants in Chinese patients causing simplex usher syndrome 2. BMC Ophthalmol. 2020;20(1):1–9.
Qu LH, Jin X, LongYL, Ren JY,Weng CH, Xu HW, Liu Y, Meng XH, Li SY, Yin ZQ. Identification of 13 novel USH2A mutations in Chinese retinitis pigmentosa and Usher syndrome patients by targeted next-generation sequencing. Biosci Rep. 2020;40(1):BSR20193536.
Richard EM, Santos-Cortez RLP, Faridi R, Rehman AU, Lee K, Shahzad M, Acharya A, Khan AA, Imtiaz A, Chakchouk I. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum Mutat. 2019;40(1):53–72.
Maranhao B, Biswas P, Gottsch AD, Navani M, Naeem MA, Suk J, Chu J, Khan SN, Poleman R, Akram J. Investigating the molecular basis of retinal degeneration in a familial cohort of Pakistani decent by exome sequencing. PLoS One. 2015;10(9):e0136561.
Li L, Chen Y, Jiao X, Jin C, Jiang D, Tanwar M, Ma Z, Huang L, Ma X, Sun W. Homozygosity mapping and genetic analysis of autosomal recessive retinal dystrophies in 144 consanguineous Pakistani families. Invest Ophthalmol Vis Sci. 2017;58(4):2218–38.
Carss KJ, Arno G, Erwood M, Stephens J, Sanchis-Juan A, Hull S, Megy K, Grozeva D, Dewhurst E, Malka S. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet. 2017;100(1):75–90.
Acknowledgements
The authors would like to thank the patients and their family members for their participation in this study.
Funding
This research was supported by the Kohat University of Science and Technology, Kohat, Pakistan.
Author information
Authors and Affiliations
Contributions
Clinical data collection, collation, and analysis: ANA, RT, MA, AB and SS; Genetic testing and data analysis: ANA, RT, MD, MY, MN, MM, KS and SS; Manuscript writing and revision: ANA, RT, MD, NK and SS; Study supervision and coordination: NK and SS. The manuscript has been read and approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval was obtained from the Ethical Committee of Kohat University of Science and Technology (KUST), and the study was carried out in accordance with the Declaration of Helsinki.
Written informed consent was obtained for participation in the study from families’ members and parents of the minor children.
Consent for publication
Written informed consent for the publication of these details have been obtained from the patient’s parents. Written consent for publication was obtained from all family members who provided DNA samples.
Competing interests
The author(s) declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ahmed, A.N., Tahir, R., Khan, N. et al. USH2A gene variants cause Keratoconus and Usher syndrome phenotypes in Pakistani families. BMC Ophthalmol 21, 191 (2021). https://doi.org/10.1186/s12886-021-01957-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-021-01957-9