
Abstract
Background
Aniridia is a kind of congenital human pan-ocular anomaly, which is related to PAX6 commonly.
Methods
The ophthalmic examinations including visual acuity, slit lamp and fundoscopy examination were performed in a Chinese aniridia pedigree. The targeted next-generation sequencing of aniridia genes was used to identify the causative mutation.
Results
A novel heterozygous PAX6 nonsense mutation c.619A > T (p.K207*) was identified in the Chinese autosomal dominant family with aniridia. Phenotype related to the novel mutation included nystagmus, keratopathy, absence of iris, cataract and foveal hypoplasia.
Conclusions
The novel nonsense variation in PAX6 was the cause of aniridia in this family, which expanded the spectrum of the PAX6 mutation.
Similar content being viewed by others
Background
Aniridia was a kind of rare congenital human ocular anomaly, it could manifest either as ocular abnormalities of iris hypoplasia, foveal hypoplasia, cataract, glaucoma, corneal dystrophy, as well as abnormalities of the optic nerve, or as syndromes of the WAGR syndrome (MIM#194,072) and the Gillespie syndrome (MIM#206,700) [1, 2]. The incidence of aniridia was between 1 and 50,000 and 96,000 births [3]. According to the difference of pathogenic genes, aniridia could be divided into three types, aniridia-1 (AN1, MIM #106,210), aniridia-2 (AN2, MIM #617,141) and aniridia-3 (AN3, MIM #617,142). AN1 was caused by heterozygous mutation in the PAX6 gene, AN2 was caused by heterozygous mutation in a PAX6 cis-regulatory element that resides in an intron of the adjacent ELP4 gene, and AN3 was caused by heterozygous mutation in the TRIM44 gene [4, 5]. Some of the isolated aniridia was demonstrated due to mutations in FOXC1 or PITX2 genes. Mutations in these two genes were more commonly associated with juvenile onset glaucoma and anterior segment dysgenesis presenting with syndromic features of rare cardiac anomalies for FOXC1 and hypodontia and umbilical anomalies for PITX2 [6,7,8].
About one-third of the cases carry de novo variants [9]. The classic AN1 associated with PAX6 haploinsufficiency presented iris hypoplasia and foveal hypoplasia, while heterozygous missense mutations in PAX6 would lead to other ocular diseases including anterior segment dysgenesis and optic nerve malformations. PAX6 (OMIM 607,108), paired box gene 6, was a member of the paired box gene family which encodes a transcriptional regulator involved in oculogenesis and other developmental processes [10]. This transcription factor had shown functional conservation in developmental pathways. PAX6 variant had been identified associated with aniridia and other ocular development abnormalities previously, while olfactory abnormalities and brain structure alterations in line with expression of Pax6 had also been documented recently [11]. PAX6 played an important role in the process of development regulation which broadly expressed and was controlled by a number of long-range control elements and homozygous mutation led to more severe phenotype [11].
In the present study, we identified a novel heterozygous PAX6 nonsense mutation c.619A > T (p.K207*) in a Chinese autosomal dominant family with aniridia. We used targeted next-generation sequencing (NGS) to screen all the genes related to iris diseases including aniridia and identified the causative mutations.
Methods
The Institutional Review Board (IRB) of Hainan hospital of Chinese PLA General Hospital (Hainan Province, China) approved the present study. All participating family members provided an informed written consent and were endorsed by their respective IRB. The whole procedure of the present study adhered to the tenets of the Declaration of Helsinki.
A small pedigree with aniridia from Hainan province, China, was recruited for the present study. This family included two affected and two unaffected members, which was analyzed and followed up clinically at Hainan Hospital of Chinese PLA General Hospital (Fig. 1a). Comprehensive ophthalmological examinations, including best correct visual acuity (BCVA), applanation tonometry, dilated funduscopy, anterior segment and fundus photography, ultrasound biomicroscopy (UBM) examination, gonioscopy and optical coherence tomography (OCT) of anterior segment and macular were performed on the affected individuals and the unaffected family members. Genomic DNA was collected from peripheral blood lymphocytes of the pedigree members and normal controls using a QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany).

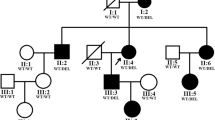
Identification of the heterozygous mutation c.619A > T in PAX6 in a Chinese family with aniridia. a Pedigree of the family. Squares indicated males and circles indicated females. Empty symbols and filled symbols represented the normal and affected individuals, respectively. Wt: wild-type and Mt: mutation. b Sequence chromatograms showing the PAX6 c.619A > T mutation identified in this study. The arrows indicated the site of the mutation
Targeted genes enrichment and sequencing
Target enrichment panel of specific hereditary eye based on the next generation sequencing was used to collect the protein-coding region of 371 targeted genes (designed by MyGenostics, Baltimore, MD), which included 37 genes associated with iris diseases. (PAX6, ELP4, FOXD3, PITX3, FOXE3, PITX2, ADAMTS10, FBN1, LTBP2, ADAMTS17, MTHFR, TYR, MITF, PAX3, SNAI2, SOX10, RBP4, CHD7, TMEM67, RPGRIP1L, CC2D2A, YAP1, MAF, C12orf57, FOXC1, B3GLCT, NF1, GPR143, OCA2, TYRP1, CRYGC, ADAMTSL4, SALL2, IGBP1, CYP1B1, MYOC, SALL1). The genes related to aniridia, WAGR syndrome, Axenfeld-Rieger syndrom and other diseases involved iris abnormity were covered. The process of specific high throughput sequencing was in conformity to some published articles [12, 13].
Bioinformatics analysis
After HiSeq 2000 sequencing, Solexa QA package and the Cutadapt program were used to filter out low quality reads and adaptor sequences and high-quality reads from raw reads were retrieved. Then the clean read sequences were aligned to the human reference genome (hg19) using SOA Paligner program. Subsequently, the single nucleotide polymorphism (SNPs) and the insertions or deletions (InDels) were identified using the SOAPsnp program and GATK program separately. The identified SNPs and InDels were annotated using ANNOVAR program (http://122.228.158.106/exomeassistant) and viewed using MagicViewer. Finally, nonsynonymous variants were evaluated by four algorithms, Ployphen, SIFT, PANTHER and Pmut, as described previously to determine pathogenicity.
Mutation verification
After high throughput sequencing, the detected variations were validated by Sanger sequencing in the Chinese family. Primer6.0 was used to design the PCR primer sets and the PCR products were sequenced using a Bigdye terminator v3.1 cycle sequencing kit (ABI, Foster City, CA, USA) and analyzed on an ABI 3730XL Genetic Analyzer. The primers used in this study are listed in Table 1.
Results
Clinical findings
There were two affected individuals, the proband and his mother, in this two-generation Chinese family (Fig. 1a). The inheritance pattern of the pedigree was in accordance with autosomal dominant inheritance. The proband (II:4) was a 29-year-old male, he felt blurred vision and photophobia in both eyes since childhood. His BCVA were 0.2 in right eyes and 0.15 in left eyes, with corrections of -4.25 diopters (D) in the right eye and − 0.5 D in the left eye. Ophthalmic examination presented horizontal nystagmus, absence of almost whole iris, discrete posterior subcapsular cataract (Fig. 2a and b), and absence of macular central fovea (Fig. 3) in both eyes. The absence of iris was so severe that the equator of the lens and ciliary processes were exposed, which could be observed in anterior segment photography (Fig. 2b and f) and UBM examination (Fig. 2e). Anterior segment photograph demonstrated that the cornea of both eyes was transparent (Fig. 2a and b), but the central corneal thickness was 668µm in right eye and 664µm in left eye (Fig. 2 c and 2d), which were slightly thicker than normal. The structure of anterior chamber angle could be observed under gonioscopy, and the anterior chamber angle was open (Fig. 2f). The normal macula central fovea structure could not be found in fundus photograph and macular OCT (Figs. 3 and 4). Intraocular pressure of both eyes was normal. His mother had similar clinical symptoms and signs, moreover, her cataract became worse with age and she underwent phacoemulsification and intraocular lens implantation at 52 years old. There were no other systemic diseases except eye abnormalities in all the affects.

Anterior segment features of the proband in the family with aniridia. a and b Slit-lamp photographs of proband. The white arrow indicated the equator of lens and the red arrows indicated discrete posterior subcapsular cataract. c and d Cornea OCT examination of proband. e Ultrasound biomicroscopy examination of proband’s right eye. (F) Anterior gonioscopy photographs. The green arrow indicated ciliary process and the orange arrow showed the structure of anterior chamber angle. OD stand for right eye, OS for left eye

Fundus examination of the proband in the family with aniridia. a and b Fundus photography of proband. The black arrows showed the structure of macular area. c and d Macular OCT examination of proband.The white arrows indicated the structure of fovea of macula. OD stands for right eye, OS, left eye

Fundus examination of the normal control. a and b Fundus photography of the normal control. The black arrows showed the structure of macular area. c and d Macular OCT examination of the normal control.The white arrows indicated the structure of fovea of macula
Identification of causing mutations
After filtering the candidate variants of the proband in the databases, a nonsense mutation PAX6 c.619A > T in exon 8 changing codon 207 AAG to the stopcodon TAG (p.K207X) was detected. The mutation was absent in either databases mentioned earlier or reported literatures, which led to Lysine at 207 position in linker region transforming to a premature termination codon, and finally resulted in PAX6 underdosage (Fig. 1b). Then the novel mutation was validated by Sanger sequencing and detected among the family members, which demonstrated that the PAX6 c.619A > T heterozygous mutation was cosegregated with the aniridia phenotype in this family (Fig. 1a). The affected individuals, the proband and his mother, carried the mutation, while the unaffected members did not. These results suggested that PAX6 c.619A > T was a novel causative mutation for autosomal dominant congenital aniridia.
Discussion
Aniridia associated with mutations in PAX6 was categorized into classic aniridia group, while that associated with mutations in genes other than PAX6 was aniridia-like group [7, 8]. Classic aniridia referred to a pan-ocular disorder, including partial or near total absence of iris, cataract, aniridia-associated keratopathy (ARK), glaucoma, foveal hypoplasia, optic disk hypoplasia and nystagmus [14,15,16]. Iris hypoplasia was the most important feature of aniridia, which could range from complete absence of the iris, through enlargement and irregularity of the pupil mimicking a coloboma, to small slit-like defects in the anterior layer seen only on transillumination with a slit-lamp. The incidence of other features of classic aniridia was different, nystagmus was 76 %, cataract was 56 %, glaucoma was 64 % and visible keratopathy was 80 % [17]. In this study, we identified the pathogenic gene mutation in a Chinese aniridia family using an iris diseases panel including 37 targeted genes. The patients in this family presented nystagmus, ARK, absence of iris, cataracts and foveal hypoplasia. Then a novel PAX6 nonsense mutation c.619A > T (p.K207*) was identified and it was co-separated from disease phenotype.
A high proportion of cases of aniridia was associated with mutations in PAX6 frameshift mutations, splicing site mutations or nonsense mutations and these kinds of variations have been considered to produce premature truncation of the protein or nonsense transcripts, leading to haploinsufficiency. While few cases were caused by missense mutations [16]. Aniridia phenotype associated with PAX6 haploinsufficiency almost present anterior segment and fundus abnormalities, while missense mutations in PAX6 were mostly associated with dysplasia of skeleton and central nervous system [18]. The most common clinical manifestations associated with aniridia haploinsufficiency were iris anomalies, nystagmus and foveal hypoplasia, followed by cataracts, glaucoma and corneal opacity. In this Chinese family, the phenotype was similar between the two patients. they both felt photophobia from childhood and exhibited nystagmus, ARK, aniridia, cataract and foveal hypoplasia. The novel mutation PAX6 c.619A > T (p.K207*) in this family induced premature termination codons (PTCs) into the PAX6 open reading frame, and the mRNAs containing PTCs were degraded by the nonsense-mediated decay process, which resulted in a single-dose deficiency. The phenotype associated with the novel mutation was in line with classic aniridia related to PAX6 haploinsufficiency [16].
It was reported that the severity of iris hypoplasia varied in different PAX6 cases and lens abnormalities include various degrees and types of cataracts and lens ectopic [16]. The patients of this Chinese family presented near total absence of iris and lamellar posterior subcapsular lens opacification without obvious lens ectopia. Keratopathy was common in aniridia patients as the PAX6 gene is responsible for embryonic and postnatal development of the cornea. At the early stage of keratopathy, the central corneal thickness increased, the basal epithelium became turbid and the corneal sensitivity decreased. With the progression of the lesion, the cornea gradually became opacity from periphery area to apex [16]. In this family, the cornea of the proband remained transparent, but the central corneal thickness increased and the limbal vascular pannus indicated the presence of early ARK. Glaucoma in aniridia usually occurred in early adulthood, including infants and toddlers. It was caused by the irregular strands arising from the iris stroma attached to the angle wall. It was reported that a 24-year-old aniridia patient with PAX6 c.607 C > T, p.Arg203* presented glaucoma, the location of the mutation was very close to that of PAX6 c.619A > T (p.K207*), and these two gene locations were in the same domain [19]. However, the patients in this Chinese aniridia family were adults with normal intraocular pressure and no characteristic optic disc manifestations of glaucoma. They need follow-up observation of intraocular pressure to finally determine whether glaucoma will occur.
The human PAX6 gene was cloned in 1991 and had been isolated from both vertebrates and invertebrates. It consisted of 14 exons and encodes a transcriptional regulator which had a paired-type DNA-binding domain. There were 2 distinct DNA-binding subdomains, the N-terminal subdomain (NTS) and the C-terminal subdomain (CTS), in the paired domain, which bind respective consensus DNA sequences to recognize target genes. The human PAX6 gene produced 2 alternatively spliced isoforms that have the distinct structure of the paired domain [20]. There have been about 500 mutations reported in the human PAX6 database [14] (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA691696), since firstly identified as the genetic cause of aniridia in the small eye mouse [21]. While it was remarkable that the alterations in a conserved non-coding element within the “critical region” and other cis-regulatory elements also could cause aniridia [22]. PAX6 whole-gene deletions and telomeric cis-regulatory elements deletions were also identified in some aniridia patients of negative for intragenic PAX6 mutations. Consequently, it was suggested that PAX6 whole-gene direct sequencing combined with and molecular methods of detecting copy number alterations (CNV) such as high-resolution comparative hybridization (HR-CGH) arrays, fluorescence in situ hybridization (FISH), and multiplex ligation-dependent probe amplification (MLPA) was important to improve detection rate for aniridia associated with PAX6 variations, which could be more suitable for using in the aniridia cases of iris diseases panel screening negative [23]. It was thought greater locus heterogeneity might exist in both isolated and syndromic aniridia than was previously appreciated, therefore the improvement of detection method was helpful to improve the detection rate of pathogenic genes for aniridia. In this study, we just have screened protein-coding region of 37 genes associated iris diseases but ignored conserved non-coding element and CNV of PAX6, which should be improved in the future.
Conclusions
In brief, a novel nonsense mutation in PAX6 (c.619A > T p.K207*) was identified in a Chinese family with aniridia, which could cause PAX6 haploinsufficiency. The phenotype associated with this mutation included aniridia, ARK, cataracts and foveal hypoplasia. The present study expanded the mutation spectrum of the PAX6 gene which may be helpful in the genetic diagnosis of aniridia.
Availability of data and materials
All data generated and analyzed during this study were included in this manuscript.
Abbreviations
- WAGR:
-
Wilms tumor, aniridia, genitourinary anomalies and mental retardation syndrome
- NGS:
-
Next-generation sequencing
- IRB:
-
Institutional review board
- BCVA:
-
Best correct visual acuity
- UBM:
-
Ultrasound biomicroscopy
- OCT:
-
optical coherence tomography
- PAX6:
-
Paired box gene 6
- ELP4:
-
Elongator acetyltransferase complex subunit 4
- FOXD3:
-
Forkhead box D3
- PITX3:
-
Paired-like homeodomain transcription factor 3
- FOXE3:
-
Forkhead box E3
- PITX2:
-
Paired-like homeodomain transcription factor 2
- ADAMTS10:
-
A disintegrin-like and metalloproteinase with thrombospondin type 1 motif, 10
- FBN1:
-
Fibrillin 1
- LTBP2:
-
Latent transforming growth factor-beta-binding protein 2
- ADAMTS17:
-
A disintegrin-like and metalloproteinase with thrombospondin type 1 motif, 17
- MTHFR:
-
Methylenetetrahydrofolate reductase
- TYR:
-
Tyrosinase
- MITF:
-
Microphthalmia-associated transcription factor
- PAX3:
-
Paired box gene 3
- SNAI2:
-
Snail family transcriptional repressor 2
- SOX10:
-
Sry-box 10
- RBP4:
-
Retinol-binding protein 4
- CHD7:
-
Chromodomain helicase dna-binding protein 7
- TMEM67:
-
Transmembrane protein 67
- RPGRIP1L:
-
Rpgrip1-like
- CC2D2A:
-
Coiled-coil and c2 domains-containing protein 2a
- YAP1:
-
Yes-associated protein 1, 65-kd
- MAF:
-
Maf bzip transcription factor
- C12orf57:
-
Chromosome 12 open reading frame 57
- FOXC1:
-
Forkhead box c1
- B3GLCT:
-
Beta-3-glucosyltransferase
- NF1:
-
Neurofibromin 1
- GPR143:
-
G protein-coupled receptor 143
- OCA2:
-
Oca2 melanosomal transmembrane protein
- TYRP1:
-
Tyrosinase-related protein 1
- CRYGC:
-
Crystallin, gamma-c
- ADAMTSL4:
-
Adamts-like 4
- SALL2:
-
Sal-like 2
- IGBP1:
-
Immunoglobulin-binding protein 1
- CYP1B1:
-
Cytochrome p450, subfamily i, polypeptide 1
- MYOC:
-
Myocilin
- SALL1:
-
Sal-like 1
- SNPs:
-
Single nucleotide polymorphism
- PCR:
-
Polymerase chain reaction
- PTC:
-
Premature termination codon
- NTS:
-
N-terminal subdomain
- CTS:
-
C-terminal subdomain
- CNV:
-
Copy number alterations
- HR-CGH:
-
High-resolution comparative hybridization
- FISH:
-
Fluorescence in situ hybridization
- MLPA:
-
Multiplex ligation-dependent probe amplification
References
Breslow NE, Takashima JR, Ritchey ML, Strong LC, Green DM. Renal failure in the Denys-Drash and Wilms’ tumor-aniridia syndromes. Cancer Res. 2000;60(15):4030–2.
Gerber S, Alzayady KJ, Burglen L, Bremond-Gignac D, Marchesin V, Roche O, Rio M, Funalot B, Calmon R, Durr A, et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am J Hum Genet. 2016;98(5):971–80. https://doi.org/10.1016/j.ajhg.2016.03.004.
Boonstra N, Limburg H, Tijmes N, van Genderen M, Schuil J, van Nispen R. Changes in causes of low vision between 1988 and 2009 in a Dutch population of children. Acta Ophthalmol. 2012;90(3):277–86. https://doi.org/10.1111/j.1755-3768.2011.02205.x.
Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T. 3’ deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci U S A. 2000;97(25):13755–9. https://doi.org/10.1073/pnas.240398797.
Zhang X, Qin G, Chen G, Li T, Gao L, Huang L, Zhang Y, Ouyang K, Wang Y, Pang Y, et al. Variants in TRIM44 Cause Aniridia by Impairing PAX6 Expression. Hum Mutat. 2015;36(12):1164–7. https://doi.org/10.1002/humu.22907.
Ito YA, Footz TK, Berry FB, Mirzayans F, Yu M, Khan AO, Walter MA. Severe molecular defects of a novel FOXC1 W152G mutation result in aniridia. Invest Ophthalmol Vis Sci. 2009;50(8):3573–9. https://doi.org/10.1167/iovs.08-3032.
Sadagopan KA, Liu GT, Capasso JE, Wuthisiri W, Keep RB, Levin AV. Anirdia-like phenotype caused by 6p25 dosage aberrations. Am J Med Genet A. 2015;167A(3):524–8. https://doi.org/10.1002/ajmg.a.36890.
Khan AO, Aldahmesh MA, Alkuraya FS. Genetic and genomic analysis of classic aniridia in Saudi Arabia. Mol Vis. 2011;17:708–14.
Vincent MC, Pujo AL, Olivier D, Calvas P. Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur J Hum Genet. 2003;11(2):163–9. https://doi.org/10.1038/sj.ejhg.5200940.
Pedersen HR, Baraas RC, Landsend ECS, Utheim OA, Utheim TP, Gilson SJ, Neitz M. PAX6 Genotypic and Retinal Phenotypic Characterization in Congenital Aniridia. Invest Ophthalmol Vis Sci. 2020;61(5):14. https://doi.org/10.1167/iovs.61.5.14.
Hever AM, Williamson KA, van Heyningen V. Developmental malformations of the eye: the role of PAX6, SOX2 and OTX2. Clin Genet. 2006;69(6):459–70. https://doi.org/10.1111/j.1399-0004.2006.00619.x.
Ying J, Lin C, Wu J, Guo L, Qiu T, Ling Y, Shan L, Zhou H, Zhao D, Wang J, et al. Anaplastic Lymphoma Kinase Rearrangement in Digestive Tract Cancer: Implication for Targeted Therapy in Chinese Population. PLoS One. 2015;10(12):e0144731. https://doi.org/10.1371/journal.pone.0144731.
Jin X, Chen L, Wang D, Zhang Y, Chen Z, Huang H. Novel compound heterozygous mutation in the POC1B gene underlie peripheral cone dystrophy in a Chinese family. Ophthalmic Genet. 2018;39(3):300–6. https://doi.org/10.1080/13816810.2018.1430239.
Lima Cunha D, Arno G, Corton M, Moosajee M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes (Basel) 2019, 10(12). https://doi.org/10.3390/genes10121050.
Wang P, Sun W, Li S, Xiao X, Guo X, Zhang Q. PAX6 mutations identified in 4 of 35 families with microcornea. Invest Ophthalmol Vis Sci. 2012;53(10):6338–42. https://doi.org/10.1167/iovs.12-10472.
Lagali N, Wowra B, Fries FN, Latta L, Moslemani K, Utheim TP, Wylegala E, Seitz B, Kasmann-Kellner B. PAX6 Mutational Status Determines Aniridia-Associated Keratopathy Phenotype. Ophthalmology. 2020;127(2):273–5. https://doi.org/10.1016/j.ophtha.2019.09.034.
Samant M, Chauhan BK, Lathrop KL, Nischal KK. Congenital aniridia: etiology, manifestations and management. Expert Rev Ophthalmol. 2016;11(2):135–44. https://doi.org/10.1586/17469899.2016.1152182.
Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E, Yamada M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003;72(6):1565–70. https://doi.org/10.1086/375555.
Dubey SK, Mahalaxmi N, Vijayalakshmi P, Sundaresan P. Mutational analysis and genotype-phenotype correlations in southern Indian patients with sporadic and familial aniridia. Mol Vis. 2015;21:88–97.
Glaser T, Walton DS, Maas RL. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992;2(3):232–9. https://doi.org/10.1038/ng1192-232.
Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V. Mouse Small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1992;355(6362):750. https://doi.org/10.1038/355750a0.
Bhatia S, Bengani H, Fish M, Brown A, Divizia MT, de Marco R, Damante G, Grainger R, van Heyningen V, Kleinjan DA. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am J Hum Genet. 2013;93(6):1126–34. https://doi.org/10.1016/j.ajhg.2013.10.028.
Ansari M, Rainger J, Hanson IM, Williamson KA, Sharkey F, Harewood L, Sandilands A, Clayton-Smith J, Dollfus H, Bitoun P, et al. Genetic Analysis of ‘PAX6-Negative’ Individuals with Aniridia or Gillespie Syndrome. PLoS One. 2016;11(4):e0153757. https://doi.org/10.1371/journal.pone.0153757.
Acknowledgements
The authors would like to thank all the patients and their family. The authors were also grateful to Dr. MR Xue and Dr. Y Wang, for they performing the ophthalmic examinations.
Funding
The funds needed for the sequencing experiment were supported by Hainan Natural Science Foundation (NO. 817347). High and New Technology Project of Hainan Key Research and Development Plan (ZDYF2020031). Special Scientific Research Project of the Logistic Support Department of the Chinese Military Committee (19BJZ39).
Author information
Authors and Affiliations
Contributions
Conceived and designed the work: HBH. Data analyzed the work: XJ, WL and LHQ. Drafted the work: XJ and WL. Substantively revised the work: XJ and WQX. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study followed the tenets of the Declaration of Helsinki and conducted with approval from the Ethics Committee of the Hainan hospital of Chinese PLA General Hospital. A written informed consent was obtained from each participant.
Consent for publication
All the patients had written informed consent for the publication of this article and all accompanying images.
Competing interests
The authors declared that they have no conflict of interest.
Data availability statement
The datasets generated during the current study are available in the https://www.ncbi.nlm.nih.gov/bioproject/PRJNA691696.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, X., Liu, W., Qv, L. et al. A novel variant in PAX6 as the cause of aniridia in a Chinese family. BMC Ophthalmol 21, 225 (2021). https://doi.org/10.1186/s12886-021-01848-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-021-01848-z