
Abstract
Background
To investigate the molecular defects in a four-generation Chinese pedigree affected with Thiel-Behnke corneal dystrophy (TBCD). And to further study the relationship between genetic mutation and clinical manifestations.
Methods
Individuals of the pedigree were recruited for extensive ophthalmic examinations. Histological studies of two corneal buttons obtained from lamellar keratoplasty were conducted. Peripheral blood was collected in EDTA for genomic DNA isolation from leukocytes of all affected and unaffected members. All 17 exons of the TGFBI gene were screened for mutations by polymerase chain reaction and direct DNA sequencing.
Results
Clinical examinations revealed a typical pattern of honeycomb-like TBCD. Histopathology study demonstrated eosinophilic deposits that were congo-red-positive and did not stain with periodic acid Schiff or Masson’s trichrome. Genetic analysis disclosed a heterozygous p. Arg555Trp mutation resulted from a missense c. 1663C > T nucleotide change in exon 12 of TGFBI gene in all affected members. Morever, a second rare variant in exon 6 of the TGFBI gene (p. Arg257Trp) also cosegregated within this family and has been confirmed to be a single nucleotide polymorphism (SNP) not previously reported.
Conclusions
The p. Arg555Trp mutation of the TGFBI gene was associated with TBCD, which revealed a novel phenotype-genotype correlation within the mutational spectrum of phenotypically diverse corneal dystrophies.
Similar content being viewed by others

Background
Corneal dystrophies (CDs) belongs to a group of inherited disorders characterized by progressive accumulation of insoluble deposits at various depths of cornea. These variable opacities often result in loss of corneal transparency and serious visual impairment [1]. Since the initial discovery by Munier et al. [2, 3] of mutations in the transforming growth factor-beta induced (TGFBI; formerly designated as BIGH3) gene located on chromosome 5q31 that cause autosomal dominant CDs, several phenotype-specific mutations have been established, which include p. Arg555Trp in granular corneal dystrophy type I (GCD1), p. Arg124His in granular corneal dystrophy type II (GCD2), p. Arg124Cys in lattice corneal dystrophy type 1 (LCD1), p. Arg555Gln in Thiel-Behnke corneal dystrophy (TBCD) and p. Arg124Leu in Reis-Bücklers corneal dystrophy (RBCD) [4]. Among these, argnine residues at positions 124 and 555 account for more than 50 % of mutations associated with TGFBI-linked CDs.
Thiel-Behnke corneal dystrophy (TBCD, OMIM 602082) was first described in 1967 which is characterized by recurrent painful corneal erosions, moderately reduced vision, autosomal dominant inheritance, and honeycomb-shaped opacities at the level of Bowman’s layer [5]. To our knowledge, TBCD was most frequently correlated with the p. Arg555Gln mutation [6, 7], and the association of p. Arg555Trp mutation with TBCD has never been reported previously. Herein, we present the clinical, histological and genetic results of a Chinese pedigree affected with TBCD and identify a novel genotype-phenotype association within TGFBI-linked CDs.
Methods
Patients recruitment and evaluation
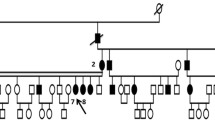
Individuals from a four-generation pedigree of Chinese ethnicity with corneal dystrophy (Fig. 1) were recruited from the Eye Center of the Second Affiliated Hospital, Medical College of Zhejiang University, Hangzhou, China. The study adhered to the tenets of the Declaration of Helsinki, and has been aproved by the ethics committee of the 2nd Affiliated Hospital of Zhejiang University.

Pedigree of the four-generation Chinese family with autosomal dominant corneal dystrophy. Squares and circles indicate males and females, respectively. Black symbols indicate affected members and open symbols indicate unaffected individuals. The diagonal line indicates a deceased family member and the black arrow indicates the proband. Asterisks indicate family members who attend this study
We received informed written consent covering DNA extraction and analysis, as well as ocular and ophthalmological examinations from all participating individuals including 150 normal controls. The written consent of the 6-year old patient for participation was obtained by her farther. Full ocular examinations including Snellen visual acuity, slit-lamp examination, applanation tonometry, corneal sensation and anterior segment photography were conducted. In addition, the patients underwent profound ophthalmologic examinations including ultrasound biomicroscopy (UBM) and anterior-segment optical coherence tomography (AS-OCT) to assess depth of the lesions.
Histological examination
Two corneal specimens of the proband were obtained from lamellar keratoplasty and underwent histopathological study. The buttons were processed by standard methods including sectioning of the tissue samples, fixation in 10 % neutral buffered formalin, then stained with hematoxylin and eosin (H&E), Congo red, Periodic acid-Schiff (PAS) and Masson’s trichrome, and analysis by light microscopy to detect the presence of deposits in the cornea.
Molecular genetic analysis
Genomic DNA was extracted from the peripheral leukocytes of 12 members (three affected and nine unaffected) according to the manufacturer’s (Simgen, Hangzhou, China) standard methods. All 17 exons of TGFBI were amplified by a polymerase chain reaction (PCR) and analyzed with the molecular method described by Zhu et al. previously [8]. PCR products were separated by electrophoresis in a 1.5 % ethidium bromide-stained agarose gel. The bands with the amplified templates were examined, and then, the purified products were sequenced according to protocols accompanying the ABI Big dye Terminator Cycle sequencing kit V 3.1 (ABI Applied Biosystems; Foster City, CA). The nucleotide sequences were analyzed by using Lasergene V. 80 software (DNASTAR, Inc., Madison, WI) and compared manually with the published NCBI TGFBI cDNA sequence (GeneBank accession no. NM. 000358). DNA from 150 normal, unrelated, ethnically Chinese volunteers (75 males and 75 females) was analyzed as the control.
Results
Phenotyping
Case III-5 (the proband)
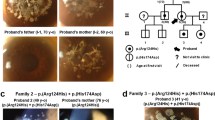
The 38-year-old male attended the outpatient clinic of our Eye Center in March 2012. He had complained of a history of recurrent, painful corneal erosions and a progressive decrease in visual acuity since he was 3 years old. Initial slit-lamp examination showed bilateral irregular corneal surfaces and discrete, dense, gray-white, honeycomb-shaped opacities in a geographic distribution involving all areas of cornea except the peripheral margin (Fig. 2). Ophthalmological exploration found a visual acuity of 0.05 in both eyes. Taken together, clinical findings support a diagnosis of TBCD. This led to bilateral lamellar keratoplasty, with an interval of five months between procedures. His Best Corrected Visual Acuity (BCVA) improved to 0.6 (OD) and 0.5 (OS) postoperatively. No episodes of rejection or recurrent dystrophic erosion have appeared thereafter with a follow-up interval of approximately 1 year.

The affected family members’ corneal phenotype illustrating TBCD as shown by slit-lamp examination. Note the tiny superficial opacities of the proband’ daughter (IV:4), bilateral irregular corneal surfaces, multiple discrete, dense gray-white, honeycomb-shaped opacities in a geographic distribution of the proband (III:5), and extensive honeycomb-shaped configuration of grayish opacities beneath the epithelium and at the Bowman’s layer of the proband’s brother (III:1). Clear area appears around the corneoscleral limbus in all the patients. These images are published with the consent of the patients, and in the case of the 6-year old patient, with the consent of her parents
Case IV-4 (the proband’ daughter)
The 6-year-old Chinese girl, was referred for intermittent attacks of ocular irritation and there was no family history of consanguinity. Her Uncorrected Visual Acuity (UCVA) was 1.0 in both eyes. Silt-lamp examination disclosed bilateral tiny superficial opacities that were prominent in whole cornea except the limbus (Fig. 2).
Case III-1 (the proband’s brother)
The 40-year-old man had complained of a very similar clinical course with his brother, and consistently showed an extensive bilateral honeycomb-shaped configuration of grayish opacities beneath the epithelium and at the Bowman’s layer (Fig. 2). Also observed were 3- to 4-mm peripheral clear zones. His BCVA was 0.12 (OD) and 0.15 (OS).
Case II-4 and Case I-1 (the proband’s father and grandfather)
The proband said these two affected patients had a history of intermittent ocular irritation, and both died blind.
Histopathologic analysis
Histopathologic analysis of the two corneal buttons from the proband revealed an irregular, layered structure of corneal epithelium in which eosinophilic deposits were noted predominantly beneath the epithelium and involved the anterior stroma within a small range by hematoxylin and eosin staining (Fig. 3a). The presence of subepithelial congo-red-positive deposits that did not stained with Periodic acid-Schiff and Masson trichrome was also illustrated (Fig. 3b, c, d). UBM and AS-OCT scan of the proband demonstrated markedly increased reflectivity, due to deposits within the lesions and irregular epithelial thickening induced by corneal opacities which project into anterior stroma (Fig. 3e, f).

Histopathologic examination of the excised corneal button from the proband. a Hematoxylin and eosin staining of the tissue section revealed an irregular, layered structure of the corneal epithelium with variable thickness in which eosinophilic deposits (asterisks) were noted predominantly beneath the epithelium and involved the anterior stroma within a small range (see the arrow). Note the obliterated Bowman’s layer and distorted epithelium with varied thickness. b Orange-coloured congo-red-positive deposits of amyloid aggregates with variable size and irregular shape are found in subepithelia area, extending into the entire stroma (arrowhead). c Numerrous small single Periodic acid Schiff (PAS) stain-negative deposits were noted throughout the corneal stroma, which disturbed the normal architecture of the stroma. d The deposits did not stain red with Masson’s trichrome. Note the apparent subepithelial fibrosis. e UBM scan demonstrated markedly increased reflectivity, due to deposits within the lesions and irregular epithelial thickening induced by corneal opacities which project into anterior stroma. f OCT-scan also confirmed an extensive sawtooth-like pattern of hyperreflective material deposited along the Bowman layer
Genotyping
Directional DNA sequencing revealed that affected members in this pedigree harbored a heterozygous missense mutation (Fig. 4), consisting of a C to T transition at nucleotide position 1663 at the first position of codon 555 in TGFBI exon 12 (c. 1663C > T), resulting in the substitution of a highly conserved Arginine by Tryptophan in the encoded TGFBIp (p. Arg555Trp). With the cosegregation of unaffected individuals and absence of this nucleotide change in 150 normal controls, we strongly assume that this mutation is pathological and exclude the possibility of its being a polymorphic change. The affected members were also heterozygous for three previously described, inconsequential, silent, single-nucleotide polymorphisms (SNPs) in exon 8, 11, 12, which did not affect the protein encoded (c.1142G > A(P.(=)), c.1577 T > C(P.(=)), c.1781C > T(P.(=)). Interestingly, in this pedigree, all patients were heterozygous for another rare variant in exon 6 of the TGFBI gene (p. Arg257Trp), (Fig. 4), which also cosegregated well within this family. We screened 150 controls of exon 6 and found 1 normal control harbored the p. Arg257Trp alteration and, thus, we considered it to be a rare SNP that was not previously reported.

DNA sequence chromatograms of the proband. Note the heterozygous C > T nucleotide change in exon 12 and 6 of TGFBI gene (see the arrow) which altered the Arg to Trp and an unaffected individual (II:5) shows Arg at the same codon 555 and 257, respectively
Discussion
Thiel-Behnke corneal dystrophy is an autosomal dominant inheritable disorder in which bilateral characteristic gray-to-white “honeycomb” corneal opacities and recurrent corneal erosions appear. It was described as corneal dystrophy affecting the surface epithelium, Bowman layer/anterior basement membrane complex and superficial stroma of the cornea by Küchle and associates [9]. Visual impairment may initially be mild, and visual acuity progressively worsens as the opacities coalesce into dense plaques over time [9, 10]. Previous studies have repeatedly shown that the missense mutation (p.Arg555Gln) of the TGFBI gene was strongly associated with TBCD [3, 7, 10–13]. In the present study, the associated of p. Arg555Trp mutation with TBCD was an unexpected and interesting finding, because, to date, the p. Arg555Trp mutation was frequently known to be responsible for subjects in corneal granular dystrophy type I (GCD I) [4, 7, 8, 11]. Therefore, our result challenges the current knowledge of “phenotypic specificity” and has permitted broadening the phenotype-moleculation correlation range within TGFBI-linked CDs.
To our knowledge, TBCD is frequently confused with Reis-Bucklers corneal dystrophy (RBCD, OMIM 608470) [14, 15], which also affects Bowman’s layer and can be difficult to differentiate both clinically and histopathologically [9, 10]. Phenotypically, this pedigree had some features that overlap with both TBCD and RBCD. Features typically associated with TBCD include honeycomb-pattern corneal opacities, relatively moderate deterioration of vision [16], and slow postoperative rate of recurrence [17], as is shown in the findings of this present study. The pathology report of subepithelial eosinophilic, congo-red-positive, PAS and Masson trichrome-negtive deposits also supported the diagnosis of TBCD [5, 18]. Meanwhile, the early disease onset occurring in the first decade of life, in keeping with RBCD. Microscopically, these two diseases can be distinguishable through transmission electron microscopy (TEM) in which RBCD is characterized by extracellular crystalloid, rod-shaped bodies [14, 15] and on the other hand, TBCD exhibits ultrastructurally “curly” fibers that stain positive for keratoepithelin [19]. Unfortunately we did not conduct TEM analysis as the tissue samples were not available, which was the limitation of our study.
What was interesting in our study is that a second heterozygous variant (p. Arg257Trp) also cosegregated with this family. Although has been confirmed to be a rare SNP, it was also predicted to change Arginine to Tryptophan in the Fas2 domain of encoded TGFBIp (NCBI Conserved Domain Database). To date, no pathogenic alterations have been found in the Fas2 region and there is no prior evidence that p. Arg257Trp alteration has any functional role, therefore, it is possible that sequence variations, especially the replacement of one residue with another, are tolerated and perhaps even modify the expression of the p. Arg555Trp mutation in this family. An effect of a yet-to-be-identified modifier genes remains a possibility. Further investigation is warranted of the mechanism of the underlying pathogenesis and an analysis of a larger cohort of families is necessary to determine whether the p. Arg257Trp polymorphism is in a linkage disequilibrium with the p. Arg555Trp mutation.
Severe visual deterioration induced by insoluble deposits of TBCD may be curable by superficial keratectomy, lamellar keratoplasty, penetrating keratoplasty (PKP), and phototherapeutic keratectomy (PTK) [20]. In our report, lamellar keratoplasty was successfully performed on the proband with a promising postoperative result.
Conclusion
In conclusion, our study identified a novel phenotype-genotype correlation between p. Arg555Trp and TBCD. The genetic, clinical, and pathological finding described here broaden the spectrum of TGFBI-linked CDs. Analysis of additional corneal dystrophy cases in different population groups will facilitated the thorough understanding of this disease.
Abbreviations
- Arg:
-
Arginine
- Trp:
-
Tryptophan
- Gln:
-
Glutamine
- His:
-
Histidine
- Cys:
-
Cysteine
- Leu:
-
Leucine
- TGFBI:
-
Transforming growth factor-beta induced
- TBCD:
-
Thiel-Behnke corneal dystrophy
- RBCD:
-
Reis-Bücklers corneal dystrophy
- SNP:
-
Single nucleotide polymorphism
- CDs:
-
Corneal dystrophies
- GCD1:
-
Granular corneal dystrophy type I
- GCD2:
-
Granular corneal dystrophy type II
- LCD1:
-
Lattice corneal dystrophy type 1
- UBM:
-
Ultrasound biomicroscopy
- AS-OCT:
-
Anterior-segment optical coherence tomography
- H&E:
-
Hematoxylin and eosin
- PAS:
-
Periodic acid-Schiff
- PCR:
-
Polymerase chain reaction
- BCVA:
-
Best Corrected Visual Acuity
- UCVA:
-
Uncorrected Visual Acuity
- TEM:
-
Transmission electron microscopy
- PTK:
-
Phototherapeutic keratectomy
References
Stone EM, Mathers WD, Rosenwasser GO, Holland EJ, Folberg R, Krachmer JH, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nat Genet. 1994;6:47–51.
Small KW, Mullen L, Barletta J, Graham K, Glasgow B, Stern G, et al. Mapping of Reis-Bucklers’ corneal dystrophy to chromosome 5q. Am J Ophthalmol. 1996;121:384–90.
Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–51.
Kannabiran C, Klintworth GK. TGFBI gene mutations in corneal dystrophies. Hum Mutat. 2006;27:615–25.
Behnke H, Thiel HJ. A previously unrecognized form of subepithelial corneal dystrophy inherited as an autosomal dominant. Ger Med Mon. 1967;12:285.
Zhao XC, Nakamura H, Subramanyam S, Stock LE, Gillette TE, Yoshikawa S, et al. Spontaneous and inheritable R555Q mutation in the TGFBI/BIGH3 gene in two unrelated families exhibiting Bowman’s layer corneal dystrophy. Ophthalmology. 2007;114:e39–46.
Munier FL, Frueh BE, Othenin-Girard P, Uffer S, Cousin P, Wang MX, et al. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci. 2002;43:949–54.
Zhu Y, Shentu X, Wang W. The TGFBI R555W mutation induces a new granular corneal dystrophy type I phenotype. Mol Vis. 2011;17:225–30.
Küchle M, Green WR, Volcker HE, Barraquer J. Reevaluation of corneal dystrophies of Bowman’s layer and the anterior stroma (Reis-Bücklers and Thiel-Behnke types): a light and electron microscopic study of eight corneas and a review of the literature. Cornea. 1995;14:333–54.
Weiss JS, Moller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, et al. IC3D classification of the corneal dystrophies-edition 2. Cornea. 2015;34:117–59.
Ridgway AE, Moller HU. Genetics of granular dystrophy. Ophthalmology. 1992;99:1753.
Chen YJ, Chen JT, Lu DW, Tai MC. In vivo corneal confocal microscopic findings and gene analysis of three patients with Thiele-Behnke corneal dystrophy. Br J Ophthalmol. 2010;94:262–4.
Mashima Y, Yamada M, Oguchi Y. Autosomal dominant inherited corneal dystrophies associated with TGFBI mutation. Nippon Ganka Gakkai Zasshi. 2001;105:659–72.
Reis W. Fämiliare, fleckige Hor nhautetartung. Dtsch Med Wochenschr. 1917;43:575 [in German].
Bücklers M. Ueber eine weitere familiar Hornhautdystrophie (Reis). Klin Monatsbl Augenheilkd. 1949;114:386–97 [in German].
Albert DM, Jakobiec FA. Principles and Practice of Ophthalmology, vol. 2. 2nd ed. Philadelphia: WB Saunders; 1994. p. 709–10.
Kamoi M, Mashima Y, Kawashima M, Tsubota K. Electrolysis for corneal opacities in a young patient with superficial variant of granular corneal dystrophy (Reis-Bücklers corneal dystrophy). Am J Ophthalmol. 2005;139:1139–40.
Ridgway AE, Akhtar S, Munier FL, Schorderet DF, Stewart H, Perveen R, et al. Ultrastructural and molecular analysis of Bowman’s layer corneal dystrophies: an epithelial origin. Invest Ophthalmol Vis Sci. 2000;41:3286–92.
Dighiero P, Niel F, Ellies P, D’Hermies F, Savoldelli M, Renard G, et al. Histologic phenotype-genotype correlation of corneal dystrophies associated with eight distinct mutations in the TGFBI gene. Ophthalmology. 2001;108:818–23.
Hieda O, Kawasaki S, Wakimasu K, Yamasaki K, Inatomi T, Kinoshita S. Clinical Outcomes of Phototherapeutic Keratectomy in Eyes With Thiel-Behnke Corneal Dystrophy. Am J Ophthalmol. 2013;155:66–72.
Acknowledgments
The authors are deeply grateful to all patients, the family, and normal volunteers for their participation in this investigation. This work was supported by the Zhejiang Key Innovation Team Project of China (No.2009R50039) and Zhejiang Key Laboratory Fund of China (No.2011E10006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KY obtained the funding, designed the research and revised the manuscript. YY carried out the molecular genetic studies, participated in the sequence alignment, analyzed most of the data, and drafted the manuscript. PQ performed the clinical examinations and operations. YZ, JL, MW carried out sample collection and helped prepare the laboratory work. BZ conducted the pathological analysis. All authors have read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yu, Y., Qiu, P., Zhu, Y. et al. A novel phenotype-genotype correlation with an Arg555Trp mutation of TGFBI gene in Thiel-Behnke corneal dystrophy in a Chinese pedigree. BMC Ophthalmol 15, 131 (2015). https://doi.org/10.1186/s12886-015-0121-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-015-0121-0