
Abstract
Background
As the most malignant tumor of the female reproductive system, ovarian cancer (OC) has garnered increasing attention. The Warburg effect, driven by glycolysis, accounts for tumor cell proliferation under aerobic conditions. However, the metabolic heterogeneity linked to glycolysis in OC remains elusive.
Methods
We integrated single-cell data with OC to score glycolysis level in tumor cell subclusters. This led to the identification of a subcluster predominantly characterized by glycolysis, with a strong correlation to patient prognosis. Core transcription factors were pinpointed using hdWGCNA and metaVIPER. A specific transcription factor regulatory network was then constructed. A glycolysis-related prognostic model was developed and tested for estimating OC prognosis with a total of 85 machine-learning combinations, focusing on specific upregulated genes of two subtypes. We identified IGF2 as a key within the prognostic model and investigated its impact on OC progression and drug resistance through in vitro experiments, including the transwell assay, lactate production detection, and the CCK-8 assay.
Results
Analysis showed that the Malignant 7 subcluster was primarily related to glycolysis. Two OC molecular subtypes, CS1 and CS2, were identified with distinct clinical, biological, and microenvironmental traits. A prognostic model was built, and IGF2 emerged as a key gene linked to prognosis. Experiments have proven that IGF2 can promote the glycolysis pathway and the malignant biological progression of OC cells.
Conclusions
We developed two novel OC subtypes based on glycolysis score, established a stable prognostic model, and identified IGF2 as the marker gene. These insights provided a new avenue for exploring OC’s molecular mechanisms and personalized treatment approaches.
Similar content being viewed by others

Introduction
Ovarian cancer (OC) is characterized by inconspicuous symptoms in its early stages and exhibits high heterogeneity, making the disease difficult to detect [1]. Among gynecological cancers (uterine, cervical, and ovarian), it has the highest mortality rate, ranking as the seventh most common malignant tumor globally, with a 49% five-year survival rate [2, 3]. Depending upon histopathology and molecular genetic characteristics, they fall into five main categories: high-grade serous (70%), endometrioid (10%), clear-cell (10%), mucinous (3%), and low-grade serous (5%) [4], each type of OC exhibits distinctive pathogenesis, immune phenotypes, and mutational landscapes. An expanding corpus of evidence underscores that OC is unique, characterized by pronounced heterogeneity induced by diverse molecular and microenvironmental factors [5,6,7]. Conventional therapeutic strategies, treating OC as a monolithic entity, are falling short in delivering targeted interventions, culminating in tumor progression, metastasis, drug resistance, and relapse, thereby impacting patient prognosis adversely [8]. Deepening insights into cancer biology have unveiled anomalous metabolic traits in cancer. Perturbed glycolysis within tumor cells, fostering glucose uptake and lactate production, fuels the metabolic processes and survival of tumor cells, thereby accelerating tumor proliferation and growth [9]. This metabolic heterogeneity further amplifies the intricacies of cancer pathophysiology [10]. Prior research has harnessed transcriptomic data to discern glycolysis-associated genes for prognostic prediction in OC patients [11]. Liu and colleagues have identified three subtypes of OC using databases and metabolic-related gene expression profiles [12].
Approximately 30% of patients experience refractory disease, displaying inherent resistance to frontline treatments, a major factor contributing to high mortality rates. High-grade serous OC (HGSOC), the most common and aggressive form of OC, constitutes approximately 70% of all cases [13]. It is usually diagnosed in advanced stages and is characterized by widespread metastases and significant ascite accumulation. Refractory cancers show inherent resistance to anti-cancer drugs before chemotherapy, which underscores the priming and protective roles of the tumor microenvironment (TME). Accordingly, there is an urgent need for comprehensive research into the underlying mechanisms and molecular profiles of OC to identify new targets and therapeutic strategies.
Glycolysis produces pyruvate under both aerobic and anaerobic conditions. Despite adequate oxygen availability, tumor-cell metabolism can be reprogrammed through glycolysis, described as the ‘Warburg effect’ in the 1920s [14]. Studies have indicated that in various cancers, including pancreatic, hepatocellular, breast, colorectal, and lung cancer, glycolysis plays a vital role in tumor invasion and growth, chemotherapy resistance, and the TME [6, 15,16,17,18,19]. Additionally, upregulated enzymes associated with glycolysis and cancer cell-induced oncogenic pathways, such as Wnt, PI3K/AKT, NF-κB, and MAPK, contribute to the extended survival and enhanced resistance of cancer cells to apoptosis [20, 21].
Glycolysis, a key driver in tumor progression, has been extensively studied to address challenges in immunotherapy. Emerging evidence suggests that the tumor immune microenvironment (TIME) considerably impacts the effectiveness and prognosis of cancer patients receiving immunotherapy [22]. Beyond its metabolic function, glycolysis induces hypoxia and lactate accumulation, affecting the TIME and immune cell function. For instance, HK2 increases PD-L1 expression in human glioblastomas through IkBa phosphorylation, thereby promoting immune evasion and cancer cell growth [23]. High-glucose conditions intensify glycolytic activity, increasing HK2 stability and impairing anti-tumor immunity in pancreatic cancer, thus accelerating disease progression [24]. Research indicates that OC cells heavily rely on glycolysis. OC cells typically show highly active glycolytic metabolism, linked to tumor invasion and recurrence [25, 26]. Thus, exploring and intervening in the glycolysis pathway is vital for understanding OC pathogenesis and developing new therapeutic approaches.
Traditional RNA sequencing is limited to systematically assessing gene expression levels and other molecular regulatory networks in biological processes. This approach has analytical limitations and fails to reveal crucial information about complex, highly heterogeneous organisms. In contrast, high-throughput single-cell RNA sequencing (scRNA-seq) represents a significant advancement over conventional sample-based experimental methods. It facilitates the detailed examination of cellular heterogeneity within and among tumors. This advanced technique can uncover individual cellular gene expression profiles, thereby exploring molecular signatures across various cell types [27, 28]. Currently, scRNA-seq is extensively being applied in various fields, such as reproduction, immunity, and the onset and development of tumors. For instance, Hao et al. conducted single-cell transcriptomic analysis of HGSOC, identifying diverse functional subclusters and molecular markers that influence OC development, thereby aiding in identifying potential treatment targets [29]. Similarly, Wang et al. utilized single-cell multi-omics sequencing to investigate features such as somatic copy number alterations, DNA methylation, and chromatin accessibility in individual cancer cells, elucidating the epigenetic and transcriptional characteristics of OC [4]. However, the mechanisms underlying metabolic heterogeneity in OC cells, particularly the influence of glycolysis-related genes in the metabolic pathway on OC prognosis and their activation in OC cells, remain insufficiently explored.
An increasing number of machine learning algorithms have been applied to various omics data analyses, including scRNAseq [30]. These algorithms efficiently train and recognize patterns in gene expression data with high accuracy, thereby reducing the time required to analyze large single-cell and spatial datasets [30, 31]. Furthermore, they also help integrate different types of biological data, offering more comprehensive biological insights [31, 32]. The insights we gained can be applied to cancer clinical management and prevention, including discovering biomarkers for early diagnosis, cancer recurrence, progression prediction, and more accurate patient survival predictions [33]. However, the performance of machine learning models relies on high-quality, large-scale annotated data, which is often challenging to obtain in bioinformatics [34]. Additionally, machine learning typically requires substantial computational resources and time. Errors in machine learning methodology can cause overfitting and overestimating performance [35]. These issues may become significant considerations in the application of machine learning and should be carefully addressed and reflected upon in our study.
This study aimed to explore the metabolic heterogeneity and diversity of OC by analyzing single-cell and transcriptomic data. We molecularly subtyped OC and analyzed the functional characteristics of each subtype, using various machine learning methods to construct a prognosis model related to glycolysis genes, thereby providing valuable insights into OC progression mechanisms and potential treatment strategies.
Materials and methods
Data source
The datasets utilized in this study are publicly accessible online. We acquired transcriptome RNA sequencing (RNA-seq) data, along with patient clinical pathological information, from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and the International Cancer Genome Consortium (ICGC, https://dcc.icgc.org/). Additionally, a dataset of single-cell sequencing for OC was downloaded from the 3CA database (https://www.weizmann.ac.il/sites/3CA/) to analyze and identify glycolysis-related OC subclusters. The glycolysis-related genes originated from the Gene Set Enrichment Analysis (GSEA) database (https://www.gsea-msigdb.org/gsea/index.jsp). The data for transcription factors was sourced from the AnimalTFDB v4.0 database (https://guolab.wchscu.cn/AnimalTFDB4/#/). Our study used data from the TCGA as the training set, and data from the ICGC as the validation set.
Sample collection
To evaluate IGF2 expression, we conducted IHC on paraffin-embedded sections from 95 patients at Shengjing Hospital of China Medical University, collected between January 2015 and December 2019. The samples included 10 normal ovarian tissues, 10 benign ovarian tumor tissues, 10 borderline ovarian tumor tissues, and 65 epithelial OC tissues. None of the patients had received radiotherapy or chemotherapy before surgery. Detailed clinicopathological and follow-up data were available for all cases in Supplementary Table 1. This study was approved by the Scientific Research and New Technology Ethical Committee of Shengjing Hospital of China Medical University, and written informed consent was obtained from all patients.
Quality control and data integration
Further quality control of cells was performed by assessing the number of detected genes (minimum: 300; maximum: 5000) and the percentage of mitochondrial genes (0–20%) for filtering purposes. Subsequently, cells were integrated using the “IntegrateData()” function from the ‘Seurat’ R package with the CCA approach to minimize technical variation between different datasets. Furthermore, criteria for inclusion in analysis: only cells that passed the quality control filters were included. Each sample included had at least 500 cells to ensure robust statistical power. The cells represented a diverse range of ovarian cancer subtypes to capture the heterogeneity of the disease.
Pseudotime analysis
We utilized the ‘Monocle3’ R package to construct single-cell pseudotime trajectories. To reduce dimensionality, the Uniform Manifold Approximation and Projection (UMAP) method was used, and the ‘plot_cells’ function was used to visualize the data. The Ucell visual scoring was employed to evaluate the activity of metabolic pathways.
Identification of specific transcription factors and their gene regulatory networks
To identify specific transcription factors and their gene regulatory networks, we constructed a scale-free network at the single-cell level using the ‘HdWGCNA’ R package. We set the scale-free topology model fitting threshold at > 0.8 and subsequently selected a soft threshold of 10 to achieve optimal connectivity. The expression levels of modules in various tumor subclusters were evaluated using Ucell visual gene scoring. To infer the activity of transcription factor proteins, we employed the PISCES process. Additionally, the Algorithm for the Reconstruction of Accurate Cellular Network (ARACNe) was utilized to construct networks that regulate transcription factor genes.
Typing construction, typing stability verification, and machine learning identification of typing marker genes
To investigate the diverse expression patterns of glycolysis-related genes in OC, we utilized 20 combination methods from the “Cola” package for clustering and classifying OC patients. For consistency in the clustering process, each iteration sampled 80% of the data, repeated 1,000 times. The optimal number of clusters (n = 2) was determined using consistency heatmap and cumulative distribution function curves. Subsequently, we employed silhouette analysis to verify the stability of the typing and the “survival” package to generate Kaplan-Meier curves, illustrating the prognosis associated with each glycolysis-related typing. The ICGC patient validation set was classified using the Nearest Template Prediction (NTP) algorithm by employing the top 200 differential genes between CS1 and CS2 as a template. To identify core marker genes for typing, we integrated 13 machine learning algorithms and 86 algorithm combinations, including Random Forest (RF), Elastic Network (Enet), NaiveBayes, Lasso, Ridge, glmBoost, LDA, Generalized Boosted Regression Modeling (GBM), plsRglm, XGBoost, ImBoost, Stepglm, and SVM. The area under the curve (AUC) was calculated to assess the model’s typing prediction stability.
Differential expression analysis of glycolysis-related genes
Based on the “limma” R package, we identified genes differentially expressed between the two molecular subtypes, CS1 and CS2. The criteria for this identification were |log2 FC| > 1 and p-value < 0.05. This analysis resulted in the identification of 83 genes.
Identification of prognostic stability and generated signature based on integrated machine learning
To develop a prognostic model with high accuracy and stability for patients with OC, we utilized differential genes identified between the CS1 and CS2 subtypes. A total of 85 machine-learning algorithm combinations were used including Enet, Lasso, Ridge, StepCox, CoxBoost, Cox partial least squares regression (plsRcox), Random Survival Forest (RSF), Supervised Principal Component (SuperPC), and GBM. The C-index was calculated for each model in both cohorts, and the model with the highest average C-index was selected as the optimal model.
Gene set enrichment analysis (GSEA)
We performed the functional analysis of the gene set using the “clusterProfiler” package in R. The gene sets, including C2.cp.kegg, C2.go.bp, and Hallmarker were sourced from the MSigDB Team.
Cell infiltration assessment
To assess cell infiltration, we employed the Cibersort algorithm, integrated with the “IOBR” package in R, to quantify the relative infiltration of 21 immune cell types in the TCGA- Ovarian Cancer (TCGA-OC) cohort. The Wilcoxon test was utilized to differentiate among the infiltrating cells. Additionally, the TME was scored using the ESTIMATE method, which evaluated stromal score, immune score, tumor purity, and a comprehensive score.
Cell lines and cell culture
The human OC cell lines, A2780 and SKOV3, were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). A2780 cells were grown in RPMI 1640 medium (Procell, Wuhan, China), supplemented with 10% fetal bovine serum (FBS; Procell), while SKOV3 cells were cultured in McCoy’s 5 A medium, supplemented with 10% FBS and 100 µg/ml streptomycin/penicillin (Gibco, Baltimore, MD, USA). The incubation procedure was carried out at 37 °C, with 5% CO2 in a moistened atmosphere.
Plasmids and transfection
The OC cells were seeded into 6-well plates. Once the cell confluence reached 50-70%, the plasmid or siRNA should be transfected using Lipo3000 (Invitrogen, USA). The medium was replaced 6 h after transfection, and the cells were continued to be cultured for subsequent experiments. Plasmids engineered to overexpress IGF2 were procured from GeneChem (Shanghai, China). The anti-IGF2 siRNA was sourced from JTSBIO Co., Ltd. (Wuhan, China). The sequences for siIGF2 were as follows: si1 (sense: 5′- CCAUGCAAAUGAAAUGUAATT − 3′; antisense: 5′- UUACAUUUCAUUUGCAUGGTT − 3′); si2 (sense: 5′- CGCAGCACAUACAUGAACATT − 3′; antisense: 5′- UGUUCAUGUAUGUGCUGCGTT − 3′).
Western blotting
Cells were lysed with RIPA lysis buffer (Beyotime, Shanghai, China), supplemented with PMSF, for 30 min. Subsequently, the lysates were centrifuged at 4 °C. The protein concentration was determined using the BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). Then, 30 µg protein from each sample was separated using 10% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA) and then blocked with 5% non-fat milk in TBST. Primary antibodies for β-actin (Proteintech, Wuhan, China, 1:20,000) and IGF2 (ABclonal, Wuhan, China, 1:500) were added and incubated overnight at 4 °C. Following washing with TBST, the membranes were incubated with rabbit IgG secondary antibody (ProteinTech) for 2 h. Immunoreactivity was detected using a Femto-sig ECL chemiluminescence kit (Tanon, Shanghai, China).
qRT-PCR
RNA was isolated with Trizol (Vazyme Biotech Co., Ltd., Nanjing, China) and subsequently reverse transcribed into cDNA with a kit (Takara Bio, Kusatsu, Japan). Quantitative PCR (qPCR) was conducted on a QuantStudio™ 5 Real-Time PCR instrument (Thermo Fisher Scientific, Waltham, MA, USA) using the SYBR Green Master Mix (Takara Bio, Kusatsu, Japan). By analyzing the threshold cycle (CT) and melting curve of the target genes, relative expression levels were calculated using the 2−ΔΔCt method. We used the following primer sequences for IGF2: forward primer: 5′-GTGGCATCGTTGAGGAGTG-3′; reverse primer: 5′-CACGTCCCTCTCGGACTTG-3′.
Immunohistochemistry (IHC)
We conducted IHC staining using an IHC kit from Maixin Biotechnology (Fuzhou, China) according to the manufacturer’s protocol. IHC analyses were performed as previously described [36]. Briefly, paraffin-embedded sections were treated with the IHC kit and then incubated overnight at 4 °C with IGF2-specific antibody (ABclonal, Wuhan, China, 1:150). The sections were then incubated with the secondary antibody from the IHC kit at 37 °C for 30 min, stained with diaminobenzidine (DAB) (ZSGB-BIO, Beijing, China), and counterstained with hematoxylin. Two independent pathologists, blinded to the experimental data, evaluated the specimens. Scoring was based on the percentage of positively stained cells (0 = 0–4%, 1 = 5–25%, 2 = 26–50%, 3 = 51–75%, 4 = 76–100%) and staining intensity (0 = none, 1 = weak, 2 = moderate, 3 = strong). The total score for each field was the product of the percentage and intensity scores.
EdU assay
Cell proliferation was evaluated using a cell proliferation assay kit (Beyotime, Shanghai, China). A complete medium enriched with the EdU solution was used to culture the cells. The cells were then fixed with 4% paraformaldehyde and then permeabilized with 0.3% Triton X-100. Subsequently, the click reaction solution was added and incubated at room temperature in the dark for 30 min. Finally, the cells were counterstained with DAPI (Beijing Solarbio Science &Technology Co., Ltd.) in the dark. Fluorescent imaging was performed using a fluorescence microscope (Nikon, Japan).
Wound healing assay
Upon transfection, the cells were seeded into six-well plates. A wound was created by scratching the cells with a 200 µl micropipette tip after they reached 100% confluence. Subsequently, the cells were gently washed with phosphate-buffered saline to remove any cellular debris and then cultured in a serum-free medium. The progress of wound healing was monitored and recorded using a phase-contrast microscope (Nikon, Japan). The area of migration was quantified using the Image J software.
Transwell assay
The transwell assay used 24-well chambers (Corning, New York, NY, USA). The upper chambers were coated with Matrigel (BD Biosciences, San Jose, CA, USA). 3 × 104 cells, suspended in 200 µL 1640 medium, were seeded into the upper chambers of each well. Meanwhile, 600 µL of 1640 medium enriched with 20% FBS were added to the lower chambers. The cells were incubated at 37 °C in a 5% CO2 environment for 24 h. After that, the cells that migrated to the lower surface of the upper chamber membrane were fixed with 4% PTA for 15 min and subsequently stained with 5% crystal violet (Beijing Solarbio Science &Technology Co., Ltd.). Following staining, photographs were taken, and the cells were counted in five random fields of view per group.
Cell counting kit-8 (CCK-8) assay
After adhering to 96-well plates at a density of 1.0 × 104 cells per well, cells were treated with varying concentrations (0, 0.5, 1, 2, 5, and 10 µg/mL) of cisplatin for 48 h. Following this, 10 µl of a suitably diluted CCK-8 solution (GlpBio Technology, Shanghai, China) was added to each well. The plates were incubated at 37 °C for 2 h before measuring the absorbance at 450 nm.
Measurement of glucose and lactate levels
The capacities of cells for glucose uptake and lactate production were evaluated using the Glucose Assay Kit (Glucose Oxidase Method) and the Lactate Assay Kit, respectively. Both kits were procured from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Thereafter, we conducted the following experiments according to the manufacturer’s instructions. After collecting cell homogenates through vortexing, proteins were quantified. Following the addition of assay reagents to the assigned groups, a reaction was initiated in a water bath maintained at 37 °C for 10 min. The absorbance of each sample was then measured.
Data analysis
All data processing, statistical analysis, and graphical plotting were performed using the R software, version 4.2.1. The relationship between two continuous variables was assessed using the Pearson correlation coefficient. For comparing continuous variables, we used either Wilcoxon’s rank-sum test or t-test, while for comparisons involving categorical variables, we used the chi-square test. The “survminer” package was used to determine optimal cut-off values for survival data. Survival analysis was conducted using the Kaplan-Meier method. Receiver Operating Characteristic (ROC) curves, crucial for predicting binary classification variables, were generated with the pROC package. Additionally, the AUC of the time-dependent ROC for survival variables was computed using the timeROC package. All statistical tests were two-sided, differences were deemed statistically significant if P < 0.05.
Results
Integration and clustering of single-cell rna sequencing data
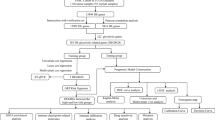
The whole flowchart of our research was outlined in Fig. 1. To enhance our understanding of OC initiation and progression, we integrated scRNA-seq data from four patients using canonical correlation analysis (CCA). Following quality control and filtering low-quality cells, we obtained a total of 15,528 cells. Subsequently, we identified six major cell types—fibroblasts (4,904), endothelial cells (616), macrophage cells (2,785), malignant cells (6,490), T cells (459), and B cells (274) (Fig. 2A). After determining 50 principal components (nPCs = 50) (Fig. 2B), we isolated malignant cells from the entire epithelial cell population. These malignant cells were then reclustered into eight distinct subclusters (Malignant 1 to Malignant 8) using UMAP for non-linear dimensionality reduction (Fig. 2C). Additionally, we identified the top five marker genes in different subclusters (Fig. 2D): Malignant 1 (CD74, NEAT1, DAPL1, HLA-DRA, and HLA-DPA1), Malignant 2 (HBB, KRT17, MEST, COL9A2, and APOA1), Malignant 3 (S100B, GAP43, FRZB, SPARCL1, and PTN), Malignant 4 (LAMC1, TGFBI, PCSK1N, TESC, and MSMP), Malignant 5 (ADIRF, NPC2, TAGLN, LCN2, and CLU), Malignant 6 (C9orf24, SAA1, TMEM190, TPPP3, and CAPS), Malignant 7 (AKAP12, DNAH5, NDUFA4L2, BNIP3, and IGF2), and Malignant 8 (RANBP1, UBE2T, CKS1B, UBE2C, and TUBA1B). We then conducted a Ucell scoring analysis to assess the activity scores of metabolic pathways in various subclusters, which revealed that Malignant 7 exhibited the highest activity in the glycolytic pathway (Fig. 2E, F). Using the ssGSEA method, we determined the optimal cutoff values for marker genes of different subclusters. This approach was then employed to identify the correlation between the Malignant 7 subcluster and the overall survival (OS) duration of patients (log-rank test, P = 0.039) (Fig. 2G).

The flowchart of our research

Single-cell interrogation of ovarian cancer revealed heterogeneity and metabolic heterogeneity. (A, B) UMAP visualization of a compendium of 15,528 single cells originating from OC, categorization was based on cell type or patient. Malignant 0: normal epithelium, Malignant 1–8: different ovarian tumor subclusters. (C) The selection of principal components was set at nPCs = 50. (D) Heatmap elucidated the top five marker genes for each tumor cell subcluster. (E) The Ucell algorithm was deployed to assess the activity of metabolic pathways across each cell subcluster. (F) Boxplot depicted the metabolic pathway activity scores across each tumor cell subcluster. (G) Kaplan-Meier survival analysis of patients in the TCGA cohort, based on the optimal cut-off value of Malignant 7 marker genes
HdWGCNA identifies hub genes of malignant 7 related to the glycolysis pathway
Subsequently, we explored the primary molecular characteristics of Malignant 7 using high-dimensional weighted gene co-expression network analysis (hdWGCNA). By setting a soft threshold of 12, we generated an optimal scale-free network for Malignant 7, facilitating the identification of gene modules (Fig. 3A, B) Simultaneously, Ucell scoring indicates that the gene module expression is most significant in Malignant 7 (Fig. 3C). Following this, we identified the three modules most relevant to Malignant 7; the top ten eigengenes of each module are displayed in Fig. 3D.

HdWGCNA identified a gene module of extreme relevance to Malignant 7. (A, B) Constructing a high-dimensional gene co-expression network. (C) The bubble chart portrayed module feature significance across different cell subclusters. (D) Presentation of the leading ten genes within three modules, arranged in order of connectivity based on respective feature genes (kME)
Establishment and verification of Oc subtypes based on glycolysis-related genes
Transcription factors are a group of protein molecules uniquely capable of binding specifically to distinct gene sequences. They ensure the expression of targeted genes at a specific intensity, within a defined timeframe, and in a precise spatial context by either suppressing or amplifying gene expression [37]. Thus, we then overlapped this transcription factor list with the module genes of the Malignant 7 group and identified 15 relevant transcription factors (HOXB9, NHLH1, NEUROG1, NEUROG1, SOX4, HOXA9, NEUROD1, SCRT2, OTP, PAX2, TFAP2B, EGR1, TCF4, ZNF536, and DACH1) (Supplementary Fig. 1A). After that, we constructed a TFs-hdWGCNA network to prognosticate the protein activity of transcription factors, among which 14 transcription factors exhibited statistically significant roles. The downstream target genes regulated by these transcription factors were also discerned and their downstream target genes (Fig. 4A).

Identification of glycolysis-related molecular subtypes in OC. (A) Construction of a TFs-hdWGCNA network to predict protein activity of the identified 15 transcription factors and to pinpoint their downstream regulated genes. (B) Utilizing twenty typing methods, the optimal number was K = 2, as indicated by the SD-skmeans method. (C) Consensus clustering differentiated two clusters, CS1 and CS2, based on glycolysis-related genes. (D) The CDF curve suggested k = 2. (E) PCA was performed using glycolysis-related genes, facilitating the separation of tumor tissue from normal tissue. (F) The stability of the subtypes was assessed using the silhouette statistical method. (G) Survival curves of OS for the consensus clusters were generated using TCGA datasets
Next, we overlapped the upregulated genes in the Malignant 7 group with the target genes of these significant transcription factors, which resulted in the identification of 440 genes (Supplementary Fig. 1B). These genes were classified using 20 different methods, then we deduced that the optimal cluster number, K = 2, with SD: skmeans emerged as the most superior method (Fig. 4B). Unsupervised consensus clustering identified two molecular subtypes, CS1 and CS2 (Fig. 4C). The cumulative distribution function curves (CDF) of the consensus score matrix indicated that the optimal cluster number was obtained when k = 2 (Fig. 4D). The proportion of ambiguous clustering (PCA) statistics further confirmed the efficient differentiation of patients into two subtypes, as visually represented in Fig. 4E. The stability of molecular clusters was assessed using silhouette statistics, and the samples were further evaluated through silhouette width. Samples with positive silhouette width were divided into two stable and robust subtypes, confirming the relative stability of the classification (Fig. 4F). To determine their clinical relevance, we further investigated the prognostic significance of these clusters. Kaplan-Meier survival estimation using samples from the TCGA database revealed that CS2 was closely associated with a poor OS, showing a low five-year survival rate (P < 0.05; Fig. 4G). Notably, CS1 exhibited more favorable survival outcomes than CS2.
Subtype analysis associated with glycolysis in OC
Using the NTP algorithm, we processed ICGC cohort data, validating the predictive confidence for each patient (Fig. 5A). Figure 5B displayed subtype ratios in TCGA and ICGC cohorts. In TCGA, CS1 was 47%, and CS2 was 53%. In contrast, ICGC had equal proportions of both subtypes at 50%, highlighting their stability. Kaplan-Meier analysis indicated that CS2 was associated with a poorer OS, whereas CS1 was associated with a better OS (P < 0.05), aligning with our previous findings (Supplementary Fig. 2A). We created models using 86 algorithms, and calculated AUC values for each model, and evaluated predictive power for ovarian cancer subtypes using TCGA and ICGC data (Fig. 5C). The Enet model achieved the highest AUC value and was selected as the final model. We once validated the AUC of this model using samples from both datasets, revealing its relatively high predictive accuracy (Supplementary Fig. 2B, C). Figure 5D illustrated the genes associated with the 86 different machine-learning methods across the two subtypes, represented as a heatmap.

Validation of the robustness of glycolysis-related ovarian cancer subtypes. (A) The heatmap illustrated the stability of OC subtypes in the ICGC database as determined by the NTP algorithm. (B) The proportions of the two subtypes were performed across the ICGC and TCGA datasets (C) A set of 86 machine-learning algorithm combinations was used to assess the stability of the typing. The AUC values of each model was computed across the ICGC and TCGA datasets. (D) Heatmap represented gene expression patterns in CS1 and CS2 as captured in the Enet[alpha = 0.5] (direction = forward) model
Underlying biological mechanisms of the subtypes
Subsequently, we conducted a differential gene analysis between CS1 and CS2 and identified 83 differential genes, including 72 upregulated and 11 downregulated genes. Figure 6A and B displayed the volcano plot and heatmap for these differential genes, respectively. The differential genes were mainly enriched in processes related to organizing extracellular structures such as the extracellular matrix (ECM) and external encapsulating structures, according to the Gene Ontology (GO) enrichment analysis (Fig. 6C). According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis, the differential genes primarily function in protein digestion and absorption and ECM-receptor interactions (Fig. 6D). Furthermore, we explored the potential biological processes and pathways of these differential genes between the two subtypes using GSEA (Fig. 6E). The results indicated significant upregulation of CS2 in various pathways, including epithelial-mesenchymal transition (EMT), mitotic spindle, UV response, hedgehog signaling, TGF beta signaling, hypoxia, apical junction, and angiogenesis. Additionally, traditional pathways such as Wnt beta-catenin signaling, G2M checkpoint, myogenesis, Notch signaling, and KRAS signaling were also notably activated in this subtype.

Elucidation of biological functional characteristics of the OC subtypes. (A) The volcano plot displayed differentially expressed genes in CS1 and CS2. Red dots indicated upregulated genes, while downregulated genes were displayed as blue dots. (B) Heatmap visualized differential gene expression in CS1 and CS2. (C, D) The biological processes of the differential genes were revealed through GO and KEGG analyses. (E) GSEA demonstrated pathway enrichment across the two subtypes
Assessment of immune infiltration and immunotherapy
In addition to the enriched pathways discussed previously, our analysis extended to the immune landscape across the two subtypes. After conducting ssGSEA analysis, we found a significant increase in infiltration abundance of various immune cells, such as resting mast cells, naive B cells, and follicular helper T cells, in CS2 compared to CS1. In contrast, activated mast cells and activated CD4(+) memory T cells were more prevalent in CS1 than in CS2 (all P < 0.05, Fig. 7A, B). Moreover, ESTIMATE analysis revealed that the stromal score was higher in CS2 than in CS1 (Fig. 7C). However, differences in ESTIMATE score, immune score, and tumor purity were minimal (Fig. 7D-F). This finding aligns with our previous enrichment analysis results, which revealed a higher level of extracellular matrix in CS2, potentially influencing patient prognosis. This suggests that patients with CS1 may be more responsive to immunotherapy than those with CS2.

Depiction of the immune landscape within the two subtypes. (A) Heatmap presented immune cell infiltration patterns across the two subtypes. (B) Analysis was performed on twenty-one types of tumor-infiltrating immune cells. (C-F) Boxplots illustrated the disparities in the stromal score, immune score, tumor purity, and ESTIMATE score across the two subtypes
Establishment of a prognostic risk model associated with the two subtypes
To construct predictive models related to glycolysis-related genes, we employed 85 modeling methods. We calculated the average C-index for each algorithm in two cohorts, ICGC and TCGA, aiming to develop the most effective model. Ultimately, the Ridge model, with the highest average C-index (0.705), was selected as the final model (Fig. 8A). We then identified IGF2, which had the highest score based on its importance to the risk model (Fig. 8B). A total of 424 patients were separated into two groups based on their IGF2 expression scores: high and low. The five-year survival rates of the two groups were compared using Kaplan-Meier curves (Fig. 8C). Our findings suggested that patients with high IGF2 expression experience shorter survival times than those with low IGF2 expression. Therefore, IGF2 can serve as a reliable prognostic indicator. Additionally, GSEA was performed to explore the correlation between IGF2 and glycolysis, revealing a positive relationship (Fig. 8D). Furthermore, enrichment analyses were conducted on the functional pathways between the two groups. In the high-IGF2 expression group, the most abundant pathways included EMT, myogenesis, apical junction, Kras signaling, and UV response (Fig. 8E). Conversely, the low-IGF2 expression group showed significant enrichment in pathways such as interferon alpha response, interferon-gamma response, allograft rejection, Myc targets V1, and oxidative phosphorylation (Fig. 8F).

Development of a prognostic model associated with glycolysis-related gene subtypes. (A) The C-index of 85 machine-learning algorithm combinations was calculated in the ICGC and TCGA databases. (B) An importance assessment was conducted for genes within the model. (C) Kaplan–Meier survival analysis compared OS between groups with high and low IGF2 expression. (D) GSEA revealed IGF2’s association with glycolysis. (E, F) GSEA enrichment analysis was performed for both the high-expression and low-expression IGF2 groups
IGF2 promoted malignant progression of OC in vitro
To investigate the expression of IGF2 in OC, we utilized information from the Human Protein Atlas (HPA) database [38]. Our analysis revealed a notable increase in IGF2 expression in OC tissues compared to normal ovarian tissues (Supplementary Fig. 3A). To compare IGF2 expression differences among various tissues, we conducted IHC analysis on 10 normal ovarian tissues, 10 benign ovarian tumor tissues, 10 borderline ovarian tumor tissues, and 65 epithelial OC tissues. The IGF2 expression scores indicated similar levels in normal and benign ovarian tumors. Compared to normal ovarian tissues, IGF2 protein levels were elevated in epithelial OC and borderline ovarian tumor tissues (Fig. 9A). Next, we analyzed the expression levels in conjunction with clinicopathological parameters (Table 1). All IHC sections from the International Federation of Gynecology and Obstetrics (FIGO) stages I-IV were derived from primary lesions. The data showed that upregulated IGF2 expression was positively correlated with higher FIGO stages. We further explored the biological function of IGF2 in OC cells by suppressing IGF2 expression in A2780 and SKOV3 cell lines using small interfering RNA and by overexpressing IGF2 using an overexpression plasmid. Following transfection, we measured IGF2 mRNA and protein levels using qRT-PCR and western blotting, respectively. Both cell lines successfully displayed IGF2 knockdown and overexpression (Fig. 9B, C; Supplementary Fig. 3B). Notably, EdU assays indicated a decrease in cell proliferation with reduced IGF2 expression, while increased IGF2 levels resulted in increased cell proliferation (Fig. 9D; Supplementary Fig. 3C). Furthermore, the assessment of wound healing and transwell assays provided corroborating evidence that the suppression of IGF2 resulted in a notable decrease in the migratory and invasive capabilities of OC cells (Fig. 9E, F). Conversely, the overexpression of IGF2 significantly augmented upon capacities (Supplementary Fig. 3D, E). Furthermore, the CCK-8 assay results demonstrated that after treatment with the specific concentration of cisplatin, the survival rate of OC cells decreased when the protein expression of IGF2 was knocked down, whereas the survival rate increased in cells with overexpression of IGF2 (Fig. 9G, Supplementary Fig. 3F). The half maximal inhibitory concentration (IC50) of A2780 and SKOV3 cells were showed in Supplementary Fig. 4A, B.

IGF2 promoted OC cell proliferation, migration, and invasion, and the resistance to cisplatin. (A) IGF2 expression in normal ovarian tissues, benign, borderline epithelial tumors, and EOC detected by IHC assay (ns, not significant; * p < 0.05; **p < 0.01,*** p < 0.001, ****p < 0.0001 in t-test. N = 5). (B) qRT-PCR analysis of IGF2 expression. (C) Western blot analysis of IGF2 expression. Protein expression levels were quantified by grey analysis (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (D) EdU assays demonstrated the cell proliferation ability upon reduction of IGF2 expression (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (E) Wound healing assays showed the migration ability of SKOV-3 and A2780 cell lines treated with si-IGF2 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (F) Transwell invasion assays showed the invasion ability of SKOV3 and A2780 cell lines treated with si-IGF2 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (G) CCK-8 assays displayed the sensitivity of OC cells to cisplatin treatment following IGF2 knockdown (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3)
IGF2 activated the glycolysis pathway
Finally, we assessed glucose uptake and lactate production in different experimental groups. Our findings showed that inhibiting IGF2 expression reduced glucose uptake and lactate production in OC cells (Fig. 10A, B), while IGF2 overexpression increased both these parameters (Supplementary Fig. 5A, B). Upon knocking down IGF2 expression, the levels of glycolysis-related proteins, including HK2, PKM2, ENO1, PGK1, and LDHA, decreased accordingly (Fig. 10C). Conversely, with overexpression of IGF2, there was an increase in the expression of proteins related to glycolysis (Supplementary Fig. 5C). After predicting the association between IGF2 and the glycolytic pathway using GSEA, we further validated experimentally that treatment with the glycolysis inhibitor 2-DG in cells overexpressing IGF2 abolished the increased proliferation, migration, and invasion capabilities of OC cells (Fig. 10D-F). Moreover, to confirm that IGF2 promoted EMT via glycolysis, western blotting analysis showed that 2-DG treatment increased the expression of the epithelial marker E-cadherin while decreasing the expression of mesenchymal markers (such as N-cadherin, Vimentin, and Snail) (Supplementary Fig. 6A). Overall, these data suggested that the activation of glycolysis was crucial for the role of IGF2 in enhancing the metabolism and invasiveness of OC cells.

IGF2 promoted the glycolysis pathway. (A) Glucose uptake experiments demonstrated the glucose uptake ability of OC cells after knockdown IGF2 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (B) Lactate production experiments indicated that IGF2 inhibition influenced lactate content generated by OC cells (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (C) Western blot analysis of glycolysis enzymes (including HK2, PKM2, ENO1, PGK1, LDHA) in IGF2-knockdown cells (The blot has been cropped, and the original blot is shown in supplementary material 7.) (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (D) Edu assay analysis revealed changes in the proliferation of OC cells with either vector control or IGF2 overexpression, treated with or without the glycolysis inhibitor 2-DG. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (E) The wound healing assay detected migration capability in vector control or IGF2-overexpressing cells, cultured with or without 2-DG. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3). (F) The transwell assay indicated the invasion capability of OC cells upon vector control or IGF2 overexpression, cultured with or without 2-DG. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in t-test. N = 3)
Discussion
As the gynecological malignancy with the highest mortality rate, OC has a notably poor prognosis and has seen no significant therapeutic breakthroughs. Prior research included establishing signatures to identify biomarkers for predicting the OC patients’ outcomes. Traditional treatment modalities often overlook the inherent heterogeneity of the disease, which may be one of the reasons for the limited effectiveness of current therapies.
Our study, for the first time, utilized single-cell data and transcriptomics to explore the heterogeneity in glycolysis of OC, revealing the metabolic characteristics and differences among various cell subclusters. The approach overcame the confounding effects presented in traditional transcriptomic studies, providing precise cellular data and capturing the unique metabolic features of different cell types within the tumor microenvironment. We have identified unique characteristics and metabolic differences between the two subtypes by molecularly profiling OC cells based on their glycolytic activity, enhancing our understanding of metabolic heterogeneity in OC. Additionally, we have developed a prognostic model based on glycolysis-related molecular subtyping and identified IGF2 as a key oncogenic factor within the model to enhance the accuracy of clinical outcome predictions and personalized treatment guidance.
Initially, by analyzing single-cell data, we identified the presence of heterogeneous subclusters within OC cells. Upon evaluation of these subclusters using metabolic pathway activity scoring, we pinpointed Malignant 5, 7, and 8 as predominantly enriched in the glycolysis pathway, suggesting activation of the glycolysis pathway during tumor progression. Notably, Malignant 7 exhibited the highest glycolysis score, indicating the highest glycolysis rate, and was significantly associated with a poor prognosis. Elevated glycolysis, a key driver of metastasis, represents an adverse phenotype in various cancers, including prostate cancer [39]. Simultaneously, it can promote resistance to cisplatin in OC, contributing to the grim prognosis of this condition [40]. These findings supported our initial observations and suggested the potential use of glycolysis inhibitors as targeted therapeutic interventions for this specific subcluster of OC cells.
In our detailed analysis of Malignant 7, we revealed its gene modules. By examining distinct transcription factors and their regulatory network genes, we categorized OC cells into two glycolysis-associated molecular subtypes, CS1 and CS2. We found that these subtypes exhibited different clinical outcomes, biological processes, and immune infiltration characteristics. Clinically, CS2 showed a more favorable prognosis than CS1. GO and KEGG enrichment analyses revealed the unique biological traits of the two subtypes. We noted a significant enrichment of differentially expressed genes in the ECM across both molecular subtypes. The ECM, a vital component of the TME, exerts considerable effects on cell survival, growth, migration, differentiation, and immune regulation [41]. The ECM undergoes continuous remodeling, driven by the synthesis and degradation of its proteins. This process is critical in tumorigenesis, as it leads to significant changes in signal transduction pathways [42], characterized by increased collagen synthesis and deposition, alongside the expression of key remodeling enzymes such as matrix metalloproteinases (MMPs) [43]. MMPs facilitate the progression of tumors by ECM proteins and enable the release of cell surface-bound cytokines, growth factors, and their receptors, which in turn promote tumor growth, invasion, metastasis, and angiogenesis [41, 44, 45]. Furthermore, the ECM in cancer creates an immunosuppressive microenvironment by modulating immune cells, which can impede the effectiveness of immune therapy [46]. Based on these observations, we hypothesized that the comparatively poorer prognosis of patients with the CS2 subtype, relative to that of those with CS1, may be due to more pronounced ECM synthesis in patients with CS2.
Next, we employed GSEA to examine the biological functional differences between the two molecular subtypes. We found that EMT, a critical tumor invasion and metastasis process, was significantly more pronounced in CS2 than in CS1 Additionally, we observed a strong enrichment of the Hedgehog and TGF-β pathways as well as angiogenesis—key processes in tissue regeneration and tumor development—in CS2. EMT is known to be a prominent feature of OC cells, playing a major role in their invasion, metastasis, and chemoresistance [47,48,49]. ECM molecules are instrumental in activating the EMT, thus facilitating the malignant progression of cancer [50]. Based on scRNA-seq analysis, a prognostic model was developed using EMT-related genes in a previous study to predict adverse patient outcomes and confirmed that cancer-associated fibroblasts promote EMT and increase tumor invasiveness [51]. Our research supported these findings and expanded upon them—the dynamic remodeling of the ECM initiates EMT, leading to a range of oncogenic signals, thus contributing to the poorer prognosis of patients in CS2.
Subsequently, from an immunological standpoint, we utilized algorithms such as ssGSEA and ESTIMATE to analyze the TIME of different subtypes. In subtype CS1, we observed increased infiltration of activated mast cells and memory CD4(+) T cells. These memory CD4(+) T cells, distributed throughout the body, play a crucial role in cell-mediated immunity, helping to eliminate tumor cells or those infected with viruses [52, 53]. Consistent with this, in a study by Liu et al., higher CD4(+) T cell density was associated with better patient outcomes [22]. Existing research has established a relationship between immune cell infiltration and the clinical prognosis of cancer patients [54]. Similar to their findings, patients with CS1 demonstrated significant immune infiltration and longer survival than those with CS1. Thus, we hypothesized that CS1 may exhibit increased sensitivity to immunotherapy, aligning with its previously observed favorable clinical prognosis. In contrast, patients with CS2 showed more significant stromal infiltration, as indicated by the presence of quiescent mast cells, natural B cells, and follicular helper T cells, compared to those in patients with CS1. As researchers have defined three immune phenotypes based on spatial distribution and intensity of T-cell infiltration, CS2 might represent the immune-desert phenotype, potentially indicating a less favorable response to immunotherapy [51].
Ultimately, to enhance the predictive accuracy, we established the model based on a combination of 85 machine learning algorithms, selecting the best model and identifying IGF2 as an important gene [20]. The IGF2 gene, located on a 150 kb locus on human chromosome 11 [55], consists of ten exons and is dynamically regulated by five distinct promoters. This allows for variable activation levels from embryonic development to postnatal life in humans [56]. IGFII, a secretory peptide comprising 67 amino acids, is encoded by the IGF2 gene. As a vital growth factor, it plays a crucial role during early embryogenesis and fetal development and is also linked to oncobiology [57,58,59]. Accumulating evidence points to the carcinogenic role of IGF2. For instance, IGF2 overexpression is often observed in adrenocortical carcinoma [60], and increased IGF2 expression in rectal cancer has been reported to be associated with poor prognosis [61]. Moreover, IGF2 not only promotes the progression of prostate cancer by inducing steroidogenesis [62] but also acts as a modulator, influencing invasive behavior in prostate cancer and cooperating with GATA2 [63]. Consistent with these observations, we observed that OC patients with high IGF2 expression tended to have a worse prognosis. IGF2 also plays significant roles in pathways such as EMT, Kras, and Hedgehog [64, 65], which suggests its potential involvement in the malignant progression of OC. Through glycolysis enrichment scoring, we further demonstrated a strong link between IGF2 and the glycolytic pathway. We hypothesized that IGF2, by activating the glycolysis pathway, induced the EMT pathway and malignant behavior in OC cells, leading to adverse outcomes and even decreased sensitivity to cisplatin and drug resistance in OC.
Subsequently, we confirmed that IGF2 enhanced the glycolytic pathway and validated its role in promoting OC cell proliferation, migration, and invasion. Additionally, using the CCK-8 assay, we demonstrated that IGF2 can reduce the effectiveness of cisplatin treatment in OC. The studies have indicated that overexpression of IGF2 in MCF7 cells results in increased proliferation, invasion, and aerobic glycolysis [66]. Moreover, OC patients with higher levels of IGF2 exhibited a poorer prognosis and shorter OS [67]. These studies collectively supported the potential of IGF2 as a therapeutic target of OC and provided a new approach for chemotherapeutic targeting, guiding personalized cancer treatment, and improving patient prognosis. To facilitate the evaluation of developing more efficient and specific IGF2 inhibitors for future screening and structural optimization, as well as validating the effectiveness of RNA interference technology in clinical trials.
There are still several limitations to consider. Firstly, these data were derived from the retrospective study using public databases, and further validation could be carried out by incorporating more advanced machine learning algorithms (such as deep learning) and conducting multicenter prospective studies [68]. Additionally, increasing the sample size can enhance the accuracy of predictions. Moreover, using CRISPR-Cas9 mediated gene knockout or knock-in experiments [69], dissecting the glycolysis-related risk model and the mechanisms behind IGF2, such as exploring the upstream and downstream molecules in the IGF2-mediated glycolytic pathway, as well as conducting organoid models for more physiologically relevant in vitro studies [70]. Finally, the study primarily focused on the glycolytic pathway, potentially overlooking the roles of other metabolic pathways or signaling pathways in tumor progression. Integrative metabolomics analysis can systematically investigate these interactions, providing a more comprehensive view of tumor metabolism.
Conclusions
Our study discovered tumor subclusters correlated with ovarian cancer prognosis based on glycolysis-related genes. Two new OC molecular subtypes, CS1 and CS2, were identified for analysis of their clinical, biological, and immune characteristics. An effective prognostic signature was conducted based on a variety of machine learning algorithms and their combinations, based on a specific transcription factor regulatory network. The key gene, IGF2, identified in the prognostic model, enhanced glycolysis and thus OC malignancy and cisplatin sensitivity.
Data availability
The transcriptome RNA-seq data from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and the International Cancer Genome Consortium (ICGC, https://dcc.icgc.org/). The dataset of single-cell sequencing for OC was downloaded from the 3CA database (https://www.weizmann.ac.il/sites/3CA/). The glycolysis-related genes originated from the Gene Set Enrichment Analysis (GSEA) database (https://www.gsea-msigdb.org/gsea/index.jsp). The data for transcription factors was sourced from the AnimalTFDB v4.0 database (https://guolab.wchscu.cn/AnimalTFDB4/#/).
Abbreviations
- OC:
-
Ovarian Cancer
- HGSOC:
-
High-Grade Serous Ovarian Cancer
- TME:
-
Tumor Microenvironment
- TIME:
-
Tumor Immune Microenvironment
- OS:
-
Overall Survival
- GSEA:
-
Gene Set Enrichment Analysis
- ROC:
-
Receiver Operating Characteristic
- AUC:
-
Area Under the Curve
- RF:
-
Random Forest
- RSF:
-
Random Survival Forest
- GBM:
-
Generalized Boosted Regression Modeling
- PAC:
-
Proportion of Ambiguous Clustering
- CT:
-
Threshold Cycle
- PVDF:
-
Polyvinylidene Difluoride
- ECM:
-
Extracellular Matrix
- HPA:
-
Human Protein Atlas
- FIGO:
-
International Federation of Gynecology and Obstetrics
- EMT:
-
Epithelial-Mesenchymal Transition
- UMAP:
-
Uniform Manifold Approximation and Projection
- CCA:
-
Canonical Correlation Analysis
- TCGA:
-
The Cancer Genome Atlas
- ICGC:
-
International Cancer Genome Consortium
- HDWGCNA:
-
High-Dimensional Weighted Gene Co-expression Network Analysis
- RNA-seq:
-
RNA Sequencing
- ssGSEA:
-
Single-Sample Gene Set Enrichment Analysis
- TGF-β:
-
Transforming Growth Factor Beta
- MMPs:
-
Matrix Metalloproteinases
- IGF2:
-
Insulin-Like Growth Factor 2
- qRT-PCR:
-
Quantitative Real-Time PCR
- CCK-8:
-
Cell Counting Kit-8
- 2-DG:
-
2-deoxy-D-glucose
- IC50:
-
Half maximal inhibitory concentration
References
Tossetta G et al. Role of NRF2 in Ovarian Cancer. Antioxid (Basel), 2022. 11(4).
Armstrong DK, et al. NCCN Guidelines® insights: ovarian Cancer, Version 3.2022. J Natl Compr Canc Netw. 2022;20(9):972–80.
Barua A, Bahr JM. Ovarian Cancer: applications of chickens to humans. Annu Rev Anim Biosci. 2022;10:241–57.
Wang Y, et al. Single-cell dissection of the Multiomic Landscape of High-Grade Serous Ovarian Cancer. Cancer Res. 2022;82(21):3903–16.
Regner MJ, et al. A multi-omic single-cell landscape of human gynecologic malignancies. Mol Cell. 2021;81(23):4924–e494110.
Sauer CM, et al. Molecular landscape and functional characterization of centrosome amplification in ovarian cancer. Nat Commun. 2023;14(1):6505.
Gong TT, et al. Proteomic characterization of epithelial ovarian cancer delineates molecular signatures and therapeutic targets in distinct histological subtypes. Nat Commun. 2023;14(1):7802.
Foster JM, et al. The contemporary management of peritoneal metastasis: a journey from the cold past of treatment futility to a warm present and a bright future. CA Cancer J Clin. 2023;73(1):49–71.
Vaupel P, Multhoff G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol. 2021;599(6):1745–57.
Arend RC, et al. Metabolic alterations and WNT signaling impact Immune Response in HGSOC. Clin Cancer Res. 2022;28(7):1433–45.
Bi J, et al. Establishment of a novel glycolysis-related prognostic gene signature for ovarian cancer and its relationships with immune infiltration of the tumor microenvironment. J Transl Med. 2021;19(1):382.
Liu X, et al. Identification of metabolism-associated molecular subtype in ovarian cancer. J Cell Mol Med. 2021;25(20):9617–26.
Wallis B et al. The challenges and prospects of p53-Based therapies in Ovarian Cancer. Biomolecules, 2023. 13(1).
Jin J, et al. Glycolysis-cholesterol metabolic axis in immuno-oncology microenvironment: emerging role in immune cells and immunosuppressive signaling. Cell Biosci. 2023;13(1):189.
Cai K, et al. FOXD1 facilitates pancreatic cancer cell proliferation, invasion, and metastasis by regulating GLUT1-mediated aerobic glycolysis. Cell Death Dis. 2022;13(9):765.
Li J, et al. CircRPN2 inhibits aerobic glycolysis and metastasis in Hepatocellular Carcinoma. Cancer Res. 2022;82(6):1055–69.
Jiang H, et al. Zeb1-induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis. 2022;13(3):206.
Yu S, et al. Deubiquitinase OTUB2 exacerbates the progression of colorectal cancer by promoting PKM2 activity and glycolysis. Oncogene. 2022;41(1):46–56.
Liu X, et al. Chromatin Remodeling Induced by ARID1A loss in Lung Cancer promotes glycolysis and confers JQ1 vulnerability. Cancer Res. 2022;82(5):791–804.
Wang L et al. Comprehensive machine-learning survival framework develops a consensus model in large-scale multicenter cohorts for pancreatic cancer. Elife, 2022. 11.
Chelakkot C et al. Modulating glycolysis to Improve Cancer Therapy. Int J Mol Sci, 2023. 24(3).
Liu C, et al. Identification of a glycolysis- and lactate-related gene signature for predicting prognosis, immune microenvironment, and drug candidates in colon adenocarcinoma. Front Cell Dev Biol. 2022;10:971992.
Guo D, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IκBα. Cell Metab. 2022;34(9):1312–e13246.
Wu S, et al. Hyperglycemia enhances immunosuppression and aerobic glycolysis of pancreatic Cancer through Upregulating Bmi1-UPF1-HK2 pathway. Cell Mol Gastroenterol Hepatol. 2022;14(5):1146–65.
Wang Z, et al. The Fibrillin-1/VEGFR2/STAT2 signaling axis promotes chemoresistance via modulating glycolysis and angiogenesis in ovarian cancer organoids and cells. Cancer Commun (Lond). 2022;42(3):245–65.
Zhang Y, et al. Metabolic dependencies and targets in ovarian cancer. Pharmacol Ther. 2023;245:108413.
Lambrechts D, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277–89.
Zhang L, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in Colon cancer. Cell. 2020;181(2):442–e45929.
Hao Q, et al. Single-cell transcriptomes reveal heterogeneity of high-grade serous ovarian carcinoma. Clin Transl Med. 2021;11(8):e500.
Dezem FS et al. A machine learning one-class logistic regression model to predict stemness for single cell transcriptomics and spatial omics. BMC Genomics, 2023. 24(1).
Marques L et al. Advancing Precision Medicine: a review of innovative in Silico approaches for Drug Development, Clinical Pharmacology and Personalized Healthcare. Pharmaceutics, 2024. 16(3).
Torun FM, et al. Transparent exploration of machine learning for Biomarker Discovery from Proteomics and Omics Data. J Proteome Res. 2022;22(2):359–67.
Mou L et al. Construction of a lipid metabolism-related risk model for hepatocellular carcinoma by single cell and machine learning analysis. Front Immunol, 2023. 14.
Cho HJ et al. Interpretable meta-learning of multi-omics data for survival analysis and pathway enrichment. Bioinformatics, 2023. 39(4).
Lakiotaki K et al. Automated machine learning for genome wide association studies. Bioinformatics, 2023. 39(9).
Wang J, et al. Regulation of ULK1 by WTAP/IGF2BP3 axis enhances mitophagy and progression in epithelial ovarian cancer. Volume 15. Cell Death & Disease; 2024. 1.
Li YF, Altman RB. Systematic target function annotation of human transcription factors. BMC Biol. 2018;16(1):4.
The Human Protein Atlas 2023; https://www.proteinatlas.org/
Ghanavat M, et al. Digging deeper through glucose metabolism and its regulators in cancer and metastasis. Life Sci. 2021;264:118603.
Papadaki C et al. Correlation of PKM2 and CD44 protein expression with poor prognosis in platinum-treated epithelial ovarian Cancer: a retrospective study. Cancers (Basel), 2020. 12(4).
Niland S, Riscanevo AX, Eble JA. Matrix metalloproteinases shape the Tumor Microenvironment in Cancer Progression. Int J Mol Sci, 2021. 23(1).
Marozzi M et al. Inflammation, Extracellular Matrix Remodeling, and Proteostasis in Tumor Microenvironment. Int J Mol Sci, 2021. 22(15).
Winkler J, et al. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11(1):5120.
Yuan Z, et al. Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. Mol Cancer. 2023;22(1):48.
Jiang Y, et al. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15(1):34.
Flies DB, et al. Regulation of tumor immunity and immunotherapy by the tumor collagen extracellular matrix. Front Immunol. 2023;14:1199513.
Loret N et al. The role of epithelial-to-mesenchymal plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers (Basel), 2019. 11(6).
Qian XQ, et al. Notch1 affects chemo-resistance through regulating epithelial-mesenchymal transition (EMT) in epithelial ovarian cancer cells. Int J Med Sci. 2020;17(9):1215–23.
Li Z, et al. Unveiling the novel immune and molecular signatures of ovarian cancer: insights and innovations from single-cell sequencing. Front Immunol. 2023;14:1288027.
Zhang J, et al. Regulation of epithelial-mesenchymal transition by tumor microenvironmental signals and its implication in cancer therapeutics. Semin Cancer Biol. 2023;88:46–66.
Xu J, et al. Single-cell RNA sequencing reveals the tissue Architecture in Human High-Grade Serous Ovarian Cancer. Clin Cancer Res. 2022;28(16):3590–602.
Liu S, et al. The relationship between CD4(+) T cell glycolysis and their functions. Trends Endocrinol Metab. 2023;34(6):345–60.
Künzli M, Masopust D. CD4(+) T cell memory. Nat Immunol. 2023;24(6):903–14.
Liu J, et al. Pyroptosis-related lncRNAs are potential biomarkers for predicting prognoses and immune responses in patients with UCEC. Mol Ther Nucleic Acids. 2022;27:1036–55.
Holthuizen P, et al. Transcriptional regulation of the major promoters of the human IGF-II gene. Mol Reprod Dev. 1993;35(4):391–3.
Sélénou C et al. IGF2: development, genetic and epigenetic abnormalities. Cells, 2022. 11(12).
Scalia P, Williams SJ, Fujita-Yamaguchi Y. Human IGF2 gene epigenetic and transcriptional regulation: at the core of Developmental Growth and Tumorigenic Behavior. Biomedicines, 2023. 11(6).
Belfiore A et al. IGF2: a role in Metastasis and Tumor Evasion from Immune Surveillance? Biomedicines, 2023. 11(1).
Scalia P, Giordano A, Williams SJ. The IGF-II-Insulin receptor Isoform-A Autocrine Signal in Cancer: actionable perspectives. Cancers (Basel), 2020. 12(2).
Scicluna P et al. Altered expression of the IGF2–H19 locus and mitochondrial respiratory complexes in adrenocortical carcinoma. Int J Oncol, 2022. 61(5).
Chatila WK, et al. Genomic and transcriptomic determinants of response to neoadjuvant therapy in rectal cancer. Nat Med. 2022;28(8):1646–55.
Lubik AA, et al. IGF2 increases de novo steroidogenesis in prostate cancer cells. Endocr Relat Cancer. 2013;20(2):173–86.
Vidal SJ, et al. A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer Cell. 2015;27(2):223–39.
Parikh K, et al. Drugging KRAS: current perspectives and state-of-art review. J Hematol Oncol. 2022;15(1):152.
Zhu C, et al. Targeting KRAS mutant cancers: from druggable therapy to drug resistance. Mol Cancer. 2022;21(1):159.
Vella V et al. Insulin receptor isoform a modulates metabolic reprogramming of breast Cancer cells in response to IGF2 and insulin stimulation. Cells, 2019. 8(9).
Hjortebjerg R et al. The IGF-PAPP-A-Stanniocalcin Axis in serum and Ascites Associates with prognosis in patients with ovarian Cancer. Int J Mol Sci, 2024. 25(4).
Kumar N, Srivastava R. Deep learning in structural bioinformatics: current applications and future perspectives. Brief Bioinform, 2024. 25(3).
Sharma AK, Giri AK. Engineering CRISPR/Cas9 therapeutics for cancer precision medicine. Front Genet, 2024. 15.
Tadić V, Zhang W, Brozovic A. The high-grade serous ovarian cancer metastasis and chemoresistance in 3D models Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 2024. 1879(1).
Acknowledgements
We would like to thank Editage for language assistance.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82272661), Liaoning key special project of science and technology (No.2022JH1/10800070), and Outstanding Scientific Fund of Shengjing Hospital (202207).
Author information
Authors and Affiliations
Contributions
J.T.J. and Q.Y.: Conceptualization; Q.Y. and J.T.J.: Methodology ; J.T.J. and F.F.B.: Software; X.C.Z. and Y.C.X.: Formal Analysis; J.T.J.: Writing – Original Draft; J.T.J., F.F.B.and Q.Y.: Writing –Review & Editing; Z.M.Z.: Visualization; Q.Y.: Supervision; Q.Y.: Funding Acquisition.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Scientific Research and New Technology Ethical Committee of Shengjing Hospital of China Medical University, and informed consent was obtained from all patients.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, J., Bi, F., Zhang, X. et al. Single-cell transcriptome analysis revealed heterogeneity in glycolysis and identified IGF2 as a therapeutic target for ovarian cancer subtypes. BMC Cancer 24, 926 (2024). https://doi.org/10.1186/s12885-024-12688-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-024-12688-7