
Abstract
Background
RAS mutations affect prognosis in patients with metastatic colorectal cancer (mCRC) and have been identified as strong negative predictive markers for anti-epidermal growth factor receptor monoclonal antibody (anti-EGFR mAb) therapy, but many tumors containing wild-type RAS genes still do not respond to these therapies. Some additional biomarkers may have prognostic or predictive roles, but conclusions remain controversial.
Methods
We performed a meta-analysis and systematic review of randomized controlled trials comparing anti-EGFR mAb therapy with alternative therapy that investigated the prognostic and predictive impact of additional biomarkers in RAS wild-type (wt) mCRC patients. Hazard ratios (HRs) and 95% confidence intervals (CIs) for progression-free survival (PFS) and overall survival (OS) and odds ratios (ORs) for objective response rate (ORR) were calculated. The prognostic value of biomarkers was investigated by separately pooling HR and OR for different treatment groups in an individual study. The predictive value was assessed by pooling study interactions between treatment effects and biomarker subgroups.
Results
Thirty publications reporting on eighteen trials were selected, including a total of 13,507 patients. In prognostic analysis, BRAF mutations were associated with poorer PFS [HRs = 3.76 (2.47–5.73) and 2.69 (1.82–3.98)] and OS [HRs = 2.66 (1.95–3.65) and 2.45 (1.55–3.88)] in both the experimental and control arms; low miR-31-3p expression appeared to have longer PFS and OS. In terms of predictive effect, a lack of response to anti-EGFR therapy was observed in patients with BRAF mutant tumors (Pinteraction < 0.01 for PFS). Patients with tumors with any mutation in the KRAS/NRAS/BRAF/PIK3CA gene also showed similar results compared with all wild-type tumors (Pinteraction for PFS, OS, and ORR were < 0.01, < 0.01 and 0.01, respectively). While low miR-31-3p expression could predict PFS (Pinteraction = 0.01) and OS (Pinteraction = 0.04) benefit. The prognostic and predictive value regarding PIK3CA mutations, PTEN mutations or deletions, EGFR, EREG/AREG, HER2, HER3, and HER4 expression remains uncertain.
Conclusions
In RAS wt mCRC patients receiving EGFR-targeted therapy, BRAF mutation is a powerful prognostic and therapy-predictive biomarker, with no effect found for PIK3CA mutation, PTEN mutation or deletion, but the combined biomarker KRAS/NRAS/BRAF/PIK3CA mutations predict resistance to anti-EGFR therapy. Low miR-31-3p expression may have positive prognostic and therapy predictive effects. Evidence on the prognostic and predictive roles of EGFR and its ligands, and HER2/3/4 is insufficient.
Similar content being viewed by others


Introduction
The epidermal growth factor receptor (EGFR) is a 170-kD transmembrane glycoprotein composed of three domains: an extracellular receptor domain, a transmembrane region, and an intracellular domain with tyrosine kinase function. It is a member of the ErbB family of receptors, a subfamily of four closely related receptor tyrosine kinases: EGFR (ErbB-1), HER2/neu (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4) [1]. Upon binding of EGF or other ligands, EGFR is activated and induces the activation of downstream signaling pathways, including Ras-MAPK, PI3K/Akt, JAK/STAT, and PLCγ/PKC pathways, which leads to tumor cell proliferation, angiogenesis, tumor invasion, metastasis, and inhibition of apoptosis [2, 3]. EGFR is overexpressed in a wide variety of solid tumors and is associated with poor prognosis [4].
Several approaches have been developed that target the EGFR to interfere with EGFR-mediated cellular effects, preventing the growth of EGFR-expressing tumors [1, 5]. The two most extensively studied to date consist of monoclonal antibodies blocking EGFR binding sites on the extracellular domain of the receptor and small-molecule compounds inhibiting intracellular tyrosine kinase activity. Anti-EGFR mAbs have been widely used in mCRC, including cetuximab (Erbitux, IMC-C225) and panitumumab (Vectibix, ABX-EGF). EGFR tyrosine kinase inhibitors (EGFR-TKIs) are effective in treating EGFR-mutated lung cancer, whereas they have thus far shown little activity in colorectal cancer [6].
The development of panitumumab and cetuximab is a milestone in the history of mCRC treatment, significantly improving the PFS and OS [7], but anti-EGFR mAb therapy is only effective for some mCRC patients [8, 9]. RAS mutations have been demonstrated to be negative predictive biomarkers of anti-EGFR mAb response and survival benefit [10, 11]. Indeed, KRAS and NRAS mutations activate downstream pathways independently of EGFR status and induce primary drug resistance. However, many tumors containing wild-type KRAS and NRAS still do not respond to these therapies, suggesting that other molecular mechanisms of resistance exist.
Several retrospective evidence suggested that mutations of BRAF, PIK3CA, loss of PTEN, aberrant expression of EGFR and its ligands amphiregulin (AREG) and epiregulin (EREG), amplification or overexpression of HER2/3, and dysregulation of microRNAs could be prognostic or predictive biomarkers of anti-EGFR mAb in RAS wt mCRC patients [12,13,14,15,16,17,18,19,20]. However, most of the conclusions are still controversial [21,22,23,24,25]. To date, only RAS and BRAF mutations have been incorporated into routine clinical practice, and the role of other biomarkers still needs to be validated. BRAF oncogene mutations are strong prognostic markers, but the predictive value for anti-EGFR mAb therapy remains a matter of debate [26, 27].
Therefore, this systematic review pooled the prognostic and predictive value of these additional biomarkers to further select patients with RAS wt mCRC who are most likely to benefit from EGFR-targeted therapy.
Methods
We performed this review according to the guidance of the Preferred Reported Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 statement [28]. The PRISMA compliance has been delineated in the PRISMA checklist table provided in Supplementary Table S1. A prospective protocol was registered in PROSPERO, CRD42022303340.
Eligibility criteria
Criteria for considering studies included: 1) Types of studies, prospective randomized clinical trials, or prospective-retrospective biomarker analysis. 2) Types of participants, RAS wt mCRC; 3) types of interventions, matched anti-EGFR mAb therapy (either as monotherapy or in combination with standard‐of‐care palliative chemotherapy) versus alternative therapy; 4) Types of outcome measures, progression‐free survival (PFS, defined as the time from trial enrolment to a composite of disease progression and death), overall survival (OS, defined as the time from trial enrolment to death from any cause), and/or overall response rate (ORR, defined as the percentage of people who achieved either a complete response or partial response) [29].
Due to the disparity in the trial protocol designs and the executing clinical centers, there were differences in the number and sort of investigated biomarkers among these studies. When we analyzed the prognostic and predictive values of each biomarker, we selected those studies containing the required data of biomarkers, which showed different numbers of studies in every analysis.
Search strategy
We systematically searched the Cochrane Library, PubMed, and Embase databases (up to 7 February 2022). The searches were rerun before the final analysis. The search strategy was presented in Additional file 1. All relevant articles were identified on PubMed to conduct a further search using the 'related articles' feature. In addition, we manually examined the citation lists of included studies and previous systematic reviews. Two authors (XL and YL) performed the search independently, in parallel.
Study selection
We imported all records retrieved by electronic searching to Endnote 20 software and removed duplicates. Three authors (YL, XZ, and JS) examined the remaining references independently, in parallel. We excluded those studies that clearly do not meet the inclusion criteria and obtained the full text of potentially relevant references. Independently, three investigators assessed the eligibility of the retrieved studies. Any disagreement was resolved through discussion or, if required, consulted a fourth person (ZY). We identified and excluded duplicate reports and collated multiple reports of the same study so that each study, rather than each report, was the unit of interest in the review.
Data extraction
Two authors (XZ, and JS) independently extracted data from included studies, resolving discrepancies by consensus or a third author (HF). Data were collected included the following: authors, publication years, journals, trial names, study design, participant demographics and characteristics, treatment protocols (lines of treatment and study treatment protocols), the status of biomarkers, outcomes (PFS, OS, and/or ORR), and results (numbers of events, hazard ratio (HR) and odds ratio (OR), and 95% confidence interval (CI). We extracted all information for data from the same trial presented in multiple publications and reported it as a single trial.
Study risk of bias assessment
We assessed and reported the risk of bias for each included study according to the Newcastle–Ottawa Scale (NOS), which is categorized into three dimensions: selection of study groups; comparability of groups; and assessment of outcomes [30, 31]. The scale for cohort studies was used because nearly all included studies were based on retrospective biomarker analysis using archived tumor specimens, and biomarker status was not a matter of randomization. Risk of bias assessments was performed independently by two authors (XL and YL) and any differences were resolved by discussion.
Statistical analysis
For prognostic and predictive analyses, PFS, OS, and ORR by treatment arm were assessed in subgroups of RAS wt patients according to the status of biomarkers. We used HRs with 95% CIs as the measure of effect for the time‐to‐event outcomes (PFS and OS). For the dichotomous outcome (ORR), OR with 95% CI was calculated. For crossover trials, we only used pre-crossover data for pooling to minimize potential bias from carry-over effects [32].
The prognostic value of biomarkers was investigated by comparing outcomes in RAS wt patients with different statuses of biomarkers using the HRs and ORs in the experimental and control arms, respectively. The predictive value of biomarkers was investigated by comparing the HRs or ORs of anti-EGFR mAb therapy (experimental arm) versus no anti-EGFR mAb therapy (control arm). Evidence for treatment effect modification by different statuses of biomarkers was evaluated by interaction tests. HRs were generally adjusted for covariates but varied to account for differences between studies. The ORs were not adjusted. The pooled HRs/ORs correspond to stratified Cox proportional hazards and logistic regression models, respectively. The HRs/ORs of interaction were pooled as proposed by Fisher et al. [33]. For data that could not be pooled statistically using meta‐analysis, we conducted a narrative synthesis of results adhering to the Synthesis Without Meta‐analysis (SWiM) guideline [34].
Heterogeneity between studies was evaluated by visual inspection of forest plots and quantified using the I2 statistic [35, 36]. I2 > 50% may represent substantial heterogeneity, in which case a random-effect (RE) model was used; otherwise, a fixed-effect (FE) model was used. The publication bias risk was assessed using funnel plots and Egger's linear regression test. Sensitivity analyses were performed to investigate the impact of excluding trials with a high risk of bias. Prespecified analyses were undertaken by grouping trials according to the anti-EGFR mAb therapy (cetuximab or panitumumab), the line of therapy, and treatment modalities in the control arm (with or without bevacizumab). All reported P values were two-sided, and all statistical analyses were carried out using R statistical software (version 4.1.2; with the meta_v5.2–0 packages).
Results
Overview of included studies and risk of bias assessment
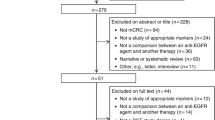
The search retrieved a total of 7658 articles that have been thoroughly reviewed for entry criteria (Fig. 1). Eighteen trials comprising 13,507 intention-to-treat (ITT) populations were finally identified that met the inclusion criteria (Table 1; Supplementary Table S2). Thirteen trials compared the addition of an anti-EGFR mAb with background treatment (FOLFIRI, FOLFOX, FLOX, irinotecan, oxaliplatin/irinotecan plus fluoropyrimidine, or best supportive care), and five compared the addition of an anti-EGFR mAb or bevacizumab to chemotherapy (FOLFOX or FOLFIRI). Twelve trials evaluated cetuximab and six assessed panitumumab. RAS mutation status was evaluable in 36%–100% of the ITT populations. Wild-type RAS accounts for approximately 59% of evaluable patients. 14 trials were available to assess the prognostic and predictive value of BRAF mutations by NGS, sanger sequencing, PCR and pyrosequencing, 4 trials for that of PIK3CA mutations by NGS, PCR and pyrosequencing, and 2 trials for that of non-functional PTEN by NGS and IHC. Three trials examined the combined effect of multiple biomarkers mutations. In addition, 8 trials assessed the value of EGFR and its ligands by IHC and PCR, 3 trials for that of other members of the HER family by PCR, and 3 trials for that of microRNA by PCR. The risk of bias assessments was summarized in Supplementary Table S3.

Flow chart of study selection
Markers downstream of EGFR
BRAF Mutations
Prognostic role of BRAF Mutations
Six trials (five panitumumab trials and one cetuximab trial) reported PFS or OS data that could be used to assess the prognostic value of BRAF mutations. For the anti-EGFR therapy arm, pooled analyses (Fig. 2a, b) showed an overall HR of 3.76 [2.47–5.73] (P < 0.01) for PFS and 2.66 [1.95–3.65] (P < 0.01) for OS in the absence of any heterogeneity between trials, indicating a negative prognostic effect of BRAF mutation. Results were similar in the control arm, with an overall HR for PFS of 2.69 [1.82–3.98] (P < 0.01; heterogeneity test P = 0.36, I2 = 1%) but less pronounced than in the experimental arm. The overall HR for OS in the control arm was 2.45 [1.55–3.88] (P < 0.01), but there was substantial heterogeneity (P < 0.01; I2 = 74%). Sensitivity analysis showed that after excluding 20,050,181 study, there was no longer significant heterogeneity for OS (P = 0.33; I2 = 12%) with an overall HR of 1.95[1.50–2.54] (P < 0.01).

Forest plots for the prognostic analyses of BRAF mutations in the control and experimental arms for progression-free survival a and overall survival b. Cmab, cetuximab; Pmab, panitumumab; mut, mutant; wt, wild-type. OS, overall survival; PFS, progression-free survival
Predictive role of BRAF Mutations
Thirteen trials reported sufficient data to assess whether the efficacy of anti-EGFR mAb differed between BRAF subgroups. PFS data were available for all 13 RCTs. Overall, the addition of anti-EGFR mAb did not increase PFS in patients with BRAF mutant tumors compared with controls [HRs of 1.05 (0.86–1.28); P = 0.62]; whereas a significant benefit of anti-EGFR mAb therapy was observed in patients with BRAF wt tumors [HRs of 0.65 (0.55–0.79); P < 0.01] (Fig. 3a). OS data were available from 11 trials except the 20,020,408 and TAILOR trials. Based on the pharmacogenomic substudies of 11 RCTs, the hazard ratio for OS benefit with anti-EGFR mAb therapy was 1.01 (0.82–1.25) for BRAF mutant tumors as compared with 0.81 (0.72–0.92) for BRAF wt tumors (Fig. 3c). Similar patterns were observed for ORR data obtained from 4 trials, with a trend toward greater benefit in the anti-EGFR mAb therapy arm in patients with BRAF wt tumors [OR = 1.93 (1.50, 2.48); P < 0.01] compared with patients with BRAF mutant tumors [OR = 1.43 (0.56, 3.64); P = 0.46] (Fig. 3e).

Forest plots for predictive analyses of BRAF mutations in trials comparing experimental arm with control arm—a, b progression-free survival, c, d overall survival and e, f objective response rate. PFS, progression-free survival; OS overall survival; ORR, objective response rate
Treatment interaction tests showed a significant difference in the PFS benefit of anti-EGFR mAb therapy between BRAF mutant and BRAF wt tumors (HR = 1.37 (1.11, 1.70), interaction test P < 0.01) (Fig. 3b). However, there was little difference was observed between BRAF mutant and BRAF wt tumors with respect to either OS efficacy (HR = 1.16 (0.93, 1.45), interaction test P = 0.40) (Fig. 3d) or ORR efficacy (OR = 0.80 (0.30, 2.13), interaction test P = 0.65) (Fig. 3f). No significant inter heterogeneity was evident between studies for the interaction test between BRAF subgroups(I2 of PFS, OS, and ORR were 26%, 4%, and 0%, respectively.). Visual inspection and linear regression tests of funnel plot asymmetry did not indicate significant publication bias.
In terms of PFS, the predictive role of BRAF status was not significantly different between studies with different anti-EGFR mAb agents (P for interaction = 0.55), different lines of therapy (P for interaction = 0.88), and with or without bevacizumab in the control arm (P for interaction = 1.00) (Table 2). However, the predictive effect was significant for studies with cetuximab, first-line therapy, or without bevacizumab (P values < 0.01, 0.02, and < 0.01, respectively) but not for studies with panitumumab, second-line and above treatment, or with bevacizumab (P values 0.21, 0.10 and 0.18, respectively). Regarding OS and ORR, no predictive effect was observed after stratification for anti-EGFR mAb used, line of treatment, and whether or not bevacizumab was included in the control arm.
PIK3CA Mutations
Prognostic role of PIK3CA Mutations
Three trials analyzed the potential prognostic value of PIK3CA mutations, but only OS data based on two trials were available for pooling. In the control arm according to PIK3CA status (mutant versus wild-type), the HR for PFS was 1.10 [0.72–1.68] (P = 0.66) for the CO.17 trial, and the pooled HR for OS was 1.11 [0.80–1.55] (P = 0.54; heterogeneity test P = 1.00, I2 = 0%) for CO.17 and PICCOLO trials. The COIN trial also showed that regardless of the treatment arm, PIK3CA mutations did not affect PFS [HR = 1.06 (0.89–1.26); P = 0.49] or OS [HR = 0.91 (0.75–1.11); P = 0.37]. This was independent of whether the PIK3CA mutation was divided into mutations in exon 9 and exon 20. Based on the small amount of trial data available, no prognostic value of PIK3CA mutations was found for patients with mCRC.
Predictive role of PIK3CA Mutations
Analysis of data based on four trials indicated that for PFS (Fig. 4a), a significant benefit of anti-EGFR mAb therapy was observed in patients with PIK3CA wt tumors [HR = 0.57 (0.38–0.87); P < 0.01], whereas no benefit was observed in patients with PIK3CA mutant tumors [HR = 0.70 (0.26–1.88); P = 0.48]; for OS (Fig. 4c), no benefit was shown in patients with both wild-type and mutant PIK3CA tumors [HRs 0.81 (0.56–1.19); P = 0.29 and 0.87 (0.49–1.52); P = 0.62, respectively]. Treatment interaction tests between PIK3CA subgroups showed no difference in the predictive value of anti-EGFR mAb therapy for both PFS (HR = 1.36 (0.89, 2.07), interaction test P = 0.15; heterogeneity test P = 0.30, I2 = 18%, Fig. 4b) and OS (HR = 1.06 (0.68, 1.65), interaction test P = 0.80; heterogeneity test P = 0.81, I2 = 0% Fig. 4d) in the absence of significant heterogeneity between studies. Stratified analysis according to the type of anti-EGFR mAb and the line of therapy also did not observe any predictive effect regarding PFS and OS (Table 3).

Forest plots for predictive analyses of PIK3CA mutations in trials comparing experimental arm with control arm—a, b progression-free survival and c, d overall survival. PFS, progression-free survival; OS overall survival
Prognostic and Predictive role of Non-functional PTEN
Here we only analyzed the data descriptively because only two trials assessed the role of PTEN mutation and reduced PTEN expression in patients with RAS wt tumors, respectively. No quantitative analysis could not be performed. For the 20,020,408 trial, a favorable effect of panitumumab on PFS was observed in patients with PTEN wt tumors [n = 135; HR = 0.36 (0.25–0.52); P < 0.001] compared with no significant benefit in patients with PTEN mutant tumors [n = 9; HR = 0.11 (0.01–1.52); P = 0.10]. The interaction term did not suggest statistical significance (HR 0.31, interaction test P = 0.36). ORRs for mutant versus wild-type PTEN among patients with wild-type KRAS who were randomized to panitumumab were 14% (95% CI, 0–0.58) and 13% (95% CI, 0.06–0.22), respectively. No patients responded to BSC alone.
For the effect of PTEN expression deficiency, the CO.17 trial showed an HR of 0.99 (P = 0.98) for PFS and 1.13 (P = 0.70) for OS between PTEN-negative and positive subgroups in the control arm, indicating no prognostic significance. Regarding predictive value, the HRs for PFS and OS between cetuximab and best supportive care were 0.66 [0.31–1.41] and 0.66 [0.29–1.52], respectively, in patients with PTEN-positive tumors and 0.34 [0.20–0.57] and 0.63 [0.38–1.03], respectively, in patients with PTEN-negative tumors. Treatment interaction tests showed no significant association between PTEN status and the survival benefit of cetuximab therapy (HR 0.52, interaction test P = 0.16 for PFS; HR 0.95, interaction test P = 0.92 for OS). The ORR to cetuximab was 21% versus 15% in patients with PTEN positive versus negative tumors. These data suggest that in patients with RAS wt tumors, PTEN status was neither prognostic nor predictive of benefit from cetuximab.
Prognostic and predictive role of multiple biomarkers
Three trials evaluated the combined impact of multiple biomarkers on clinical outcomes of anti-EGFR therapy in mCRC. The role of BRAF and PIK3CA mutations in patients with RAS wt was analyzed in the VISNÚ-2 trial, which showed similar trends in PFS improvement in patients with BRAF/PIK3CA wt versus BRAF- and/or PIK3CA-mutated tumors in both treatment arms [FOLFIRI plus cetuximab: HR = 1.55 (0.83–2.89); P = 0.17; FOLFIRI plus bevacizumab: HR = 1.07 (0.61- 1.88); P = 0.83]. The predictive effect of chemotherapy plus EGFR antibody therapy compared with chemotherapy plus bevacizumab was not significantly different for patients with BRAF/PIK3CA wt and BRAF- or PIK3CA-mutated tumors both for PFS (HR 1.12, interaction test P = 0.79), OS (HR 0.87, interaction test P = 0.73), and ORR (OR 0.71, interaction test P = 0.65).
Two additional trials assessed whether the efficacy of anti-EGFR mAb differed between KRAS, NRAS, BRAF, and PIK3CA combined biomarker subgroups. The pooled analyses showed that significant PFS and ORR benefits of anti-EGFR mAb therapy were observed in patients with all-wt tumors [HR 0.66 (0.53–0.82); P < 0.01, OR 5.32 (3.16–8.96); P < 0.01] compared with no benefit in those with mutant (any mutation in KRAS/NRAS/BRAF/PIK3CA genes) tumors [HR 1.32 (0.97–1.81); P = 0.08, OR 1.41 (0.63–3.18); P = 0.41] (Fig. 5a, e). The results for OS showed a similar trend (Fig. 5c), with significantly shorter OS in patients with any mutant tumors [HR 1.63 (1.20, 2.22); P < 0.01] compared to patients with all-wild type tumors [HR 0.78 (0.50, 1.22); P = 0.28]. Treatment interaction tests showed that the predictive value of anti-EGFR mAb therapy was significantly different for patients with mutant and wild-type KRAS/NRAS/BRAF/PIK3CA tumors all for PFS (HR = 2.01 (1.36, 2.96), interaction test P < 0.01; heterogeneity test P = 0.24, I2 = 29%, Fig. 5b), OS (HR = 1.96 (1.34, 2.86), interaction test P < 0.01; heterogeneity test P = 0.34, I2 = 0%, Fig. 5d) and ORR (OR = 0.27 (0.10, 0.74), interaction test P = 0.01; heterogeneity test P = 0.61, I2 = 0%, Fig. 5f).

Forest plots for predictive analyses of multiple biomarkers in trials comparing experimental arm with control arm—a, b progression-free survival, c, d overall survival and e, f objective response rate. PFS, progression-free survival; OS overall survival; ORR, objective response rate
EGFR and its ligands
Prognostic and predictive role of EGFR
Because of the significant heterogeneity of EGFR status, detection methods, and determination criteria in different studies, no quantitative synthesis of data was performed. Three trials used immunohistochemistry to assess the correlation between EGFR immunostaining and clinical response to anti-EGFR mAb therapy. The COIN trial showed a prognostic effect of EGFR on PFS at the standardized cutoff point of < 10% vs. ≥ 10% [HR 1.25 (1.05–1.50); P = 0.015] in the KRAS wt cohort (Table 4). There was no evidence of EGFR immunohistochemistry's predictive role, regardless of the cutoff point (data not shown). Data from the CRYSTAL and TAILOR studies also confirm that adding cetuximab to chemotherapy improved the survival benefit of first-line treatment for patients with RAS wt mCRC, irrespective of tumor EGFR status (Fig. 6a-c).

Forest plots for predictive analyses of EGFR in trials comparing experimental arm with control arm—a progression-free survival, b overall survival and c objective response rate. NA, not available; PFS, progression-free survival; OS overall survival; ORR, objective response rate
The CALGB 80203 trial reported data from patients with EGFR expression evaluated by quantitative polymerase chain reaction and no prognostic association was found between this parameter and survival outcomes in KRAS wt patients [HRs of 0.80 (0.58–1.09); P = 0.168 and 1.04 (0.81–1.34); P = 0.748 for PFS and OS, respectively]. EGFR gene expression levels were also not a potential predictive marker for cetuximab benefit (HR 0.74, interaction test P = 0.37 for PFS; and HR 0.88, interaction test P = 0.86 for OS).
Two trials (the 20,020,408 and PICCOLO trials) provided data from EGFR amplification patients, showing that patients with increased EGFR gene copy number had higher response rates and longer PFS to anti-EGFR mAb compared to patients with normal EGFR gene copy number (Table 4; Fig. 6a). Data from the 20,020,408 trial showed that in patients treated with panitumumab, the mean EGFR GCN ≥ 2.5/nucleus or percentage of chromosome 7 polysomy ≥ 40% predicted better PFS (P = 0.039 and 0.029, respectively) and OS (P = 0.015 and 0.014, respectively). Six of 20 patients with EGFR GCN ≥ 2.47/nucleus and six of 19 patients with chromosome 7 polysomy ≥ 43% achieved an objective response ((P = 0.0009 and 0.0007, respectively). In contrast, no patients had tumor response when the EGFR GCN or chromosome 7 polysomy was less than this value. In this trial, there was no correlation between EGFR GCN and chromosome 7 polysomy status and PFS in patients receiving supportive care, suggesting that this parameter is not prognostic in mCRC. The PICCOLO trial showed a similar trend, with no prognostic effect of EGFR copy number gain on PFS (P = 0.98) and OS (P = 0.97). However, it was predictive of panitumumab benefit, with median PFS of 5.7 vs 3.7 months in RAS wt patients with EGFR-gain [HR 0.60 (0.43–0.83), P = 0.002], but no benefit in patients with normal EGFR [3.4 vs 2.9 months, HR 1.23 (0.72–2.08); P = 0.45) (HR 0.49, interaction test P = 0.02). In RAS wt patients, EGFR gain was associated with higher response rates than normal in the irinotecan plus panitumumab arm (45.3% vs. 18.7%, P = 0.01) but not in the irinotecan arm (13.3% vs. 12.9%, P = 1.0) (Table 4). The interaction was not significant (P = 0.22).
Prognostic and Predictive role of EGFR Ligands
Five trials evaluated the EGFR ligands EREG and AREG as prognostic and predictive biomarkers. We did not quantitatively synthesize the data due to the apparent differences between studies.
In terms of EREG/AREG as a combined dichotomous biomarker, data from the PICCOLO trial confirmed that high ligand mRNA levels or IHC positivity are predictive biomarkers of benefit from panitumumab treatment in patients with metastatic colorectal cancer (Fig. 7a-c). In RAS wt patients with high ligand mRNA levels (either ligand in the top tertile), panitumumab treatment had a significantly longer PFS compared with control treatment [HR 0.38 (0.24–0.61); P < 0.001]. However, there was no benefit of panitumumab in RAS wt patients with low ligand mRNA levels (neither ligand in top tertile) [HR, 0.93 (0.64–1.37); P = 0.73]. The ligand-treatment interaction was significant (HR 0.41, interaction test P = 0.01). The effects on OS (HR 0.64, interaction test P = 0.11) and ORR (OR 3.69, interaction test P = 0.088) were less significant. The trial also explored several alternative cutpoints, including the 50th, 80th, and 90th centiles, but none separated the beneficiary/non-beneficiary population to a greater extent. Analysis of immunohistochemistry showed similar results, with high ligand IHC positivity (> 50% AREG or > 50% EREG) associated with significant PFS and ORR benefit with panitumumab [HR for PFS 0.54 (0.37–0.79); P = 0.001 and OR for ORR 14.1 (4.58, 43.39)); P = 0.000]; and no benefit in patients with low ligand IHC positivity (≤ 50% AREG and ≤ 50% EREG) [HR for PFS 1.05 (95% CI, 0.74–1.49); P = 0.78, and OR for ORR 2.07 (0.87, 4.91); P = 0.10]. Treatment interaction tests were significant both for PFS (HR 0.51, interaction test P = 0.02) and ORR (OR 6.81, interaction test P = 0.008). The results for OS were less significant (HR 0.72, interaction test P = 0.19). The effects of different cutoffs were also examined here and found that interactions remained significant across the 20% to 50% cutoff range. For the prognostic role of tumor EREG/AREG expression in RAS wt patients, no prognostic effect of high versus low expresser status on PFS or OS was seen in the subgroup treated with irinotecan alone (Table 5).

Forest plots for predictive analyses of EGFR ligands in trials comparing experimental arm with control arm—a progression-free survival, b overall survival and c objective response rate. NA, not available; PFS, progression-free survival; OS overall survival; ORR, objective response rate
The trials also examined EREG and AREG separately as independent biomarkers. For EREG, contradictory results were presented between these studies. When examined as a dichotomous variable, in the prognostic analysis, EREG expression had a favorable prognostic effect on OS in the control arm [HR 0.79 (0.66–0.94)] in the CALGB 80203 trial but failed to show significance in the CO.17 and PICCOLO trials (Table 5). In predictive analysis, the CO.17 trial showed that patients with high EREG expression appeared to benefit more from cetuximab treatment than those with low expression (HR 0.49, interaction test P = 0.04 for OS, Fig. 7b), and the PICCOLO trial also found that the percentage of EREG IHC positivity > 50% predicted PFS (HR 0.50, interaction test P = 0.02, Fig. 7a) and OS (HR 0.60, interaction test P = 0.04, Fig. 7b) benefits of panitumumab. However, no predictive value was observed in the CALGB 80203 and COIN trials. As a continuous variable, a prognostic impact of EGFR expression on OS was observed in both the CO.17 and PICCOLO trials [HR 0.85 (0.96–0.76); P = 0.01 and 0.87 (0.80–0.94); P = 0.001, respectively] (Table 5), but did not predict survival benefit of anti-EGFR mAb therapy. In contrast, in the immunohistochemical analysis of the PICCOLO trial, EGFR IHC positivity was not prognostic for either PFS or OS in patients treated with irinotecan only but predicted a PFS benefit with panitumumab (HR 0.91, interaction test P = 0.01, Table 5). EREG status may have certain prognostic and predictive roles, but no definitive conclusions can be drawn based on current evidence.
When AREG was analyzed separately as a dichotomous variable, the CALGB 80203 and PICCOLO trials showed that AREG mRNA expression and AREG IHC were neither prognostic markers nor predictive markers of benefit from EGFR-targeted therapy in RAS wt metastatic colorectal cancer (Table 5; Fig. 7a, b). When examined as a continuous variable, neither AREG expression nor AREG IHC had prognostic significance for PFS and OS. In the predictive analysis of patients with RAS wt tumors, AREG expression predicted the effect of panitumumab treatment on PFS (HR 0.82, interaction P = 0.008) but not OS (HR 0.89, interaction test P = 0.07); and AREG IHC was not predictive of either PFS (HR 0.91, interaction test P = 0.06) or OS (HR 0.96, interaction test P = 0.43) (Table 5).
Prognostic and Predictive role of other members of the HER family
Data on HER2 and HER4 were only available from the CALGB 80203 trial (Table 6). Prognostic analysis showed no significant effect of HER2 and HER4 expression on PFS and OS in both the anti-EGFR therapy and control groups. The interaction test between treatment and gene expression also did not show a predictive value.
For HER3, neither of the two included trials (the PICCOLO and CALGB 80203 trials) observed evidence for it as a prognostic biomarker but found tumor HER3 mRNA expression may be a useful predictive biomarker for anti-EGFR therapy in RAS wt patients (Table 6; Fig. 8a-c). However, the prediction of HER3 expression levels in these two trials was in opposite directions. Considering the substantial statistical heterogeneity (I2 = 88%) associated with methodological and clinical characteristics, we ultimately decided not to perform a quantitative synthesis of the trials because it would be clinically meaningless, and the results would be difficult to interpret. In the CALGB 80203 trial, high HER3 expression predicted a lack of OS benefit from cetuximab therapy [chemotherapy plus cetuximab: HR 1.15 (0.81–1.62); chemotherapy alone: HR 0.48 (0.27–0.87); interaction test P = 0.029] (Table 6). Conversely, in the PICCOLO trial, patients with high HER3 mRNA expression significantly benefited from panitumumab, both as a continuous variable and a binary model. There was a significant interaction between biomarkers and treatment (continuous variable: HR 0.78, interaction test P = 0.003 for PFS; HR 0.81, interaction test P = 0.01 for OS, Table 6; dichotomous variable: HR 0.34, interaction test P = 0.002 for PFS; HR 0.42, interaction test P = 0.01 for OS, Fig. 8a, b).

Forest plots for predictive analyses of HER3 in trials comparing experimental arm with control arm—a progression-free survival, b overall survival and c objective response rate. NA, not available; PFS, progression-free survival; OS overall survival; ORR, objective response rate
Prognostic and Predictive role of MicroRNA
Three trials assessed the value of relevant microRNAs as potential biomarkers. In the PICCOLO trial, a predefined model classified RAS wt patients (n = 188) into three tertiles, high, intermediate (int), and low miR-31-3p expression. Int and high expression patients had worse PFS (HR 1.60, 1.60; P = 0.018, respectively) and OS (HR 1.58, 2.03; P = 0.0012, respectively) compared with low expression patients and after adjustment for the treatment arm (Table 7), indicating a positive prognostic effect of low miR-31-3p expression. In predictive analysis, panitumumab produced a significant PFS benefit in patients with low and int miR-31-3p expression (HR 0.50; P = 0.019 and 0.57; P = 0.031, respectively) but not in patients with high expression (HR 0.72; P = 0.23) (Fig. 9a); however, no statistically significant treatment-expression interaction.

Forest plots for predictive analyses of miR-31-3p in trials comparing experimental arm with control arm—a, d progression-free survival, b, e overall survival and c, f objective response rate. NA, not available; PFS, progression-free survival; OS overall survival; ORR, objective response rate
Data from the FIRE-3 trial not only showed the prognostic value of miR-31-3p expression but also observed a significant benefit of anti-EGFR mAb therapy in patients with low miR-31-3p expression tumors compared with no benefit in patients with high miR-31-3p expression tumors, but none of these results from the New EPOC trial were statistically significant (Table 7; Fig. 9a-c). Pooled results from two trials confirmed that miR-31-3p expression levels predicted PFS (HR = 1.83 (1.15, 2.93), interaction test P < 0.01; heterogeneity test P = 0.80, I2 = 0%, Fig. 9d) and OS (HR = 1.81 (1.02, 3.21), interaction test P = 0.04; heterogeneity test P = 0.86, I2 = 0%, Fig. 9e) benefits of anti-EGFR mAb therapy, but not for ORR (OR = 0.63 (0.17, 2.30), interaction test P = 0.48; heterogeneity test P = 0.14, I2 = 53%, Fig. 9f).
In addition, a study of patients from the FIRE-3 trial also suggested that miR-21 expression levels may be a predictive biomarker for anti-EGFR therapy (data not shown).
Discussion
Colorectal cancer accounts for about one-tenth of global cancer and death cases, ranking third in incidence but second to lung cancer in terms of mortality [65]. Metastatic colorectal cancer is associated with a significantly worse prognosis, with a 5-year relative survival rate of no more than 15% for patients [66]. Since the US FDA approved cetuximab for the treatment of mCRC in 2004, EGFR-targeted therapy has become an essential means to improve the survival prognosis of RAS wt mCRC patients. However, primary and secondary resistance prevent many patients from benefiting from this therapy. Therefore, identifying other potential molecular biomarkers to guide this treatment and prognostic stratification of mCRC patients is highly desirable. We identified 30 publications in this systematic review involving 18 well-known RCTs, providing a comprehensive exploration of potential biomarkers currently widely studied.
The first to be extensively evaluated were critical components of the EGFR signaling pathway, including RAS, BRAF, PIK3CA, PTEN, and combinations of them [67]. This is driven by plausible biological rationale that constitutive activation of signaling pathways parallel to or downstream of EGFR should circumvent EGFR inhibition and therefore preclude sensitivity to anti-EGFR mAbs [68]. Some previous meta-analyses have highlighted the prognostic value of BRAF mutations, but there is insufficient evidence to demonstrate its predictive role for anti-EGFR mAbs therapy [26, 69, 70]. The efficacy of anti-EGFR mAb in patients with BRAF-mutated mCRC is still under debate. Recently, preliminary results from the randomized phase II FIRE-4.5 study (AIO KRK-0116) provided good data [71]. This is the first trial to investigate mFOLFOXIRI in combination with cetuximab or bevacizumab as first-line treatment for patients with RAS wild-type, BRAF V600E mutant mCRC. According to the results of the FIRE-4.5 trial presented at the 2021 ASCO Annual meeting, FOLFOXIRI plus cetuximab (49.2%) did not induce a higher ORR compared with FOLFOXIRI plus bevacizumab (60.0%); and both PFS and OS were significantly better in the bevacizumab group than in the cetuximab-treated group. This coincides with our results. Our pooled analysis of relevant data from 14 trials determined that BRAF mutation was not only a negative prognostic biomarker in patients with RAS wt tumors but also predicted a lack of benefit from anti-EGFR mAb therapy (interaction test P < 0.01 for PFS). Stratified analysis showed that the predictive effect of BRAF status on PFS might depend on cetuximab, first-line treatment, and the absence of bevacizumab in the control group. Although some studies have initially suggested that RAS wt mCRC patients with PIK3CA mutations and PTEN mutations or deletions may have lower responses and poorer outcomes when receiving anti-EGFR mAbs [72,73,74,75,76]. But we found no prognostic or predictive role for them based on the data from several available trials. However, a pooled analysis of two trials evaluating the combined biomarkers of KRAS, NRAS, BRAF, and PIK3CA showed a significantly reduced benefit from anti-EGFR therapy in patients with any mutant tumor compared with patients with all wt tumors (interaction tests P < 0.05 for PFS, OS, and ORR). This suggests that combinatorial analysis of multiple biomarkers can help further screen outpatient populations that may benefit.
EGFR and its ligands EREG and AREG are commonly overexpressed in colorectal cancer [77]. Autocrine stimulation of EGFR by AREG and EREG is a mechanism of tumor EGFR pathway dependence, so the impact of their expression on the response to EFGR-targeted therapy in mCRC patients has also received much attention [78,79,80]. Several retrospective studies have suggested that EGFR status and EREG/AREG expression may correlate with the prognosis of mCRC patients and may identify patients who will or will not benefit from anti-EGFR therapy [16, 80,81,82,83,84,85,86]. However, the strength of the evidence is limited, and the conclusions are controversial, so further validation of the clinical utility of these markers is needed [23]. Under this topic, we only performed a descriptive analysis of relevant trial data because of the high heterogeneity among studies. EGFR was targeted in six trials involving its immunohistochemical protein expression, mRNA expression, and frequency of gene amplification. No prognostic association has been found between this parameter and survival outcomes in RAS wt patients. In terms of predictive effects, data from the 20,020,408 and PICCOLO trials suggest that patients with increased EGFR GCN had higher response rates to panitumumab and longer PFS; however, there was no evidence of a predictive role for EGFR protein and mRNA expression. Four trials analyzed EREG and AREG alone or as a combined biomarker. For EREG/AREG as a combined dichotomous biomarker, high ligand mRNA levels or IHC positivity could predict PFS and ORR benefit from panitumumab treatment, but no prognostic effect was observed. When EREG and AREG were investigated as separate biomarkers, conflicting results emerged, and EREG expression may have certain prognostic and predictive roles compared with AREG. Still, no clear conclusions can be drawn at this time.
The HER family of receptor tyrosine kinases shares a high degree of structural and functional homology, which is the molecular basis for receptor interaction and cross-activation [87]. Dimers containing HER3, especially the HER2-HER3 heterodimer, have been shown to act as oncogenic units to drive tumor cell proliferation [88]. And preclinical experiments showed that cetuximab induces HER2-HER3 dimers in colon cancer cells [89].Several small clinical reports support the importance of expression of other markers in the HER axis, suggesting that HER2 amplification and HER3 overexpression appear to be predictive markers of resistance to anti-EGFR mAb therapy [90,91,92,93,94]. Of the included trials, only two provided data on HER2, HER3, and HER4 expression. In prognostic analysis, no meaningful results were observed for all three. In terms of predictive effect, only tumor HER3 mRNA expression may be a predictive biomarker for anti-EGFR therapy in RAS wt patients. However, in contrast to the results of previous retrospective studies and the CALGB 80203 trial, the PICCOLO trial showed that HER3 overexpression was significantly associated with benefit rather than resistance to anti-EGFR therapy. This result may be due to the role of HER3 as an obligate heterodimer. Overall, further studies are needed to verify the role of other members of the HER family.
MicroRNAs (miRNAs) are a group of short non-coding RNAs that play important roles in carcinogenesis and tumor progression, and their aberrant expression may potentially be used as diagnostic, therapeutic, and prognostic markers for CRC [95,96,97,98]. MiR-31 is frequently upregulated in CRC tumors compared with normal mucosa and miR-31 activates the RAS pathway and functions by repressing RAS p21 GTPase activating protein 1 translation, thereby promoting CRC progression [99, 100]. In addition, it has been shown that low expression of miR-31-3p could be a consequence of the regulation of pre-mir-31 maturation by an EGFR-activated pathway, driving tumor sensitivity to anti-EGFR therapy [58].Our meta-analysis showed that low miR-31-3p expression predicted PFS (interaction test P = 0.01) and OS benefit (interaction test P = 0.04) from anti-EGFR mAb treatment. In terms of prognostic analysis, low miR-31-3p expression also showed a positive prognostic effect. In addition, a trial assessing the expression level of miR-21 also showed preliminary predictive value.
To our knowledge, this is the first systematic review to systematically summarize the evidence from RCTs on the prognostic and predictive value of all extensively studied potential biomarkers of interest, and the largest on this topic. However, the current analysis also has some limitations that must be acknowledged. First, the results of the trials we included in the analysis were extracted from published data and not based on individual patient data. Second, some biomarkers were only analyzed descriptively with limited strength of evidence due to limitations in the number of included trials and the high heterogeneity caused by differences in assay methods, determination criteria, etc. Furthermore, stratified analyzes of interest could not be performed in each marker due to the limited number of studies. Moreover, some factors that may influence the impact of mutations on survival, such as MSI status, were limited by the fact that the original report did not adjust the hazard ratio for them, and could not be further analyzed. Finally, some of the studies included in the analysis were only reported as abstracts in conference presentations rather than fully published articles, and results may differ slightly between future full publications.
Conclusions
In conclusion, our analysis was able to confirm that, in RAS wt mCRC patients, BRAF mutations were associated with poor prognosis and predicted lack of response to anti-EGFR therapy, and combination biomarker KRAS/NRAS/BRAF/PIK3CA mutations were also negative predictive markers for treatment; low miR-31-3p expression was positive prognostic and predictive of therapy. There is currently insufficient evidence to support PIK3CA mutations, PTEN mutations or deletions, EGFR status, and HER2 and HER4 expression as prognostic or therapy predictive biomarkers. EREG/AREG and HER3 expression may have a particular predictive role, but the conclusions are still controversial. These results are preliminary, and efforts are needed to achieve assay standardization and prospective validation to optimize further the identification of patients who will benefit from anti-EGFR therapy.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
References
Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):21–6.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37.
Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. 2005;2005(1):0010.
Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9-15.
Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051–8.
Santoro A, Comandone A, Rimassa L, Granetti C, Lorusso V, Oliva C, et al. A phase II randomized multicenter trial of gefitinib plus FOLFIRI and FOLFIRI alone in patients with metastatic colorectal cancer. Ann Oncol. 2008;19(11):1888–93.
Stintzing S, Modest DP, Rossius L, Lerch MM, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): a post-hoc analysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol. 2016;17(10):1426–34.
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25(13):1658–64.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–45.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–34.
Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol. 2015;26(1):13–21.
Santos C, Azuara D, Garcia-Carbonero R, Alfonso PG, Carrato A, Elez ME, et al. Optimization of RAS/BRAF Mutational Analysis Confirms Improvement in Patient Selection for Clinical Benefit to Anti-EGFR Treatment in Metastatic Colorectal Cancer. Mol Cancer Ther. 2017;16(9):1999–2007.
Sato T, Osumi H, Shinozaki E, Ooki A, Shimozaki K, Kamiimabeppu D, et al. Clinical impact of primary tumor location and RAS, BRAF V600E, and PIK3CA mutations on epidermal growth factor receptor inhibitor efficacy as third-line chemotherapy for metastatic colorectal cancer. Anticancer Res. 2021;41(8):3905–15.
Tural D, Batur S, Erdamar S, Akar E, Kepil N, Mandel NM, et al. Analysis of PTEN, BRAF and PI3K status for determination of benefit from cetuximab therapy in metastatic colorectal cancer patients refractory to chemotherapy with wild-type KRAS. Tumour Biol. 2014;35(2):1041–9.
Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19(7):1902–12.
Randon G, Yaeger R, Hechtman JF, Manca P, Fucà G, Walch H, et al. EGFR amplification in metastatic colorectal cancer. J Natl Cancer Inst. 2021;113(11):1561–9.
Kim SA, Park H, Kim KJ, Kim JW, Sung JH, Nam M, et al. Amphiregulin can predict treatment resistance to palliative first-line cetuximab plus FOLFIRI chemotherapy in patients with RAS wild-type metastatic colorectal cancer. Sci Rep. 2021;11(1):23803.
Takahashi N, Yamada Y, Furuta K, Honma Y, Iwasa S, Takashima A, et al. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2014;110(11):2716–27.
Huang W, Chen Y, Chang W, Ren L, Tang W, Zheng P, et al. HER2 positivity as a biomarker for poor prognosis and unresponsiveness to anti-EGFR therapy in colorectal cancer. J Cancer Res Clin Oncol. 2022;148(4):993–1002.
Aksoy S, Kanat O, Tunca B, Ertas H, Ugras N, Ozturk E, et al. MicroRNA dysregulation as a prognostic biomarker in wild-type RAS/RAF/PTEN/PI3 metastatic colon cancer treated with anti-EGFR therapeutics. Ann Oncol. 2018;29:iii30.
Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15(9):3184–8.
Tol J, Dijkstra JR, Klomp M, Teerenstra S, Dommerholt M, Vink-Börger ME, et al. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer. 2010;46(11):1997–2009.
Klupp F, Sass M, Bergmann F, Khajeh E, Ghamarnejad O, Hassenpflug M, et al. Impact of EGFR and EGFR ligand expression on treatment response in patients with metastatic colorectal cancer. Oncol Lett. 2021;21(6):448.
Stahler A, Heinemann V, Neumann J, Crispin A, Schalhorn A, Stintzing S, et al. Prevalence and influence on outcome of HER2/neu, HER3 and NRG1 expression in patients with metastatic colorectal cancer. Anticancer Drugs. 2017;28(7):717–22.
Boisteau E, Lespagnol A, De Tayrac M, Corre S, Perrot A, Rioux-Leclercq N, et al. MiR-31-3p do not predict anti-EGFR efficacy in first-line therapy of RAS wild-type metastatic right-sided colon cancer. Clin Res Hepatol Gastroenterol. 2022;46(5): 101888.
Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer. 2015;112(12):1888–94.
Loree JM, Dowers A, Tu D, Jonker DJ, Edelstein DL, Quinn H, et al. Expanded low allele frequency RAS and BRAF V600E testing in metastatic colorectal cancer as predictive biomarkers for cetuximab in the randomized CO.17 trial. Clin Cancer Res. 2021;27(1):52–9.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372: n71.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47.
Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010;25(9):603–5.
Wells GA, Shea B, O’Connell D, Peterson J, Welch V, Losos M, et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. Oxford; 2000.
Higgins J, Li T, Deeks J. Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor (s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www training cochrane org/handbook. 2021.
Fisher DJ, Copas AJ, Tierney JF, Parmar MK. A critical review of methods for the assessment of patient-level interactions in individual participant data meta-analysis of randomized trials, and guidance for practitioners. J Clin Epidemiol. 2011;64(9):949–67.
Campbell M, McKenzie JE, Sowden A, Katikireddi SV, Brennan SE, Ellis S, et al. Synthesis without meta-analysis (SWiM) in systematic reviews: reporting guideline. BMJ. 2020;368: l6890.
Deeks JJ. Statistical methods for examining heterogeneity and combining results from several studies in meta-analysis. Systematic reviews in health care. 2001:285–312.
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–60.
Sartore-Bianchi A, Moroni M, Veronese S, Carnaghi C, Bajetta E, Luppi G, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25(22):3238–45.
Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, et al. Analysis of KRAS/NRAS mutations in a phase III study of Panitumumab with FOLFIRI Compared with FOLFIRI alone as second-line treatment for metastatic colorectal cancer. Clin Cancer Res. 2015;21(24):5469–79.
Kim TW, Elme A, Park JO, Udrea AA, Kim SY, Ahn JB, et al. Final analysis of outcomes and RAS/BRAF status in a randomized phase 3 study of panitumumab and best supportive care in chemorefractory wild type KRAS metastatic colorectal cancer. Clin Colorectal Cancer. 2018;17(3):206–14.
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48(10):1466–75.
Licitra L, Störkel S, Kerr KM, Van Cutsem E, Pirker R, Hirsch FR, et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: analysis of data from the EXTREME and CRYSTAL studies. Eur J Cancer. 2013;49(6):1161–8.
Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O’Callaghan CJ, Marginean C, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer--results from NCIC CTG/AGITG CO.17. Clin Cancer Res. 2014;20(3):744–53.
Jonker DJ, Karapetis CS, Harbison C, O’Callaghan CJ, Tu D, Simes RJ, et al. Epiregulin gene expression as a biomarker of benefit from cetuximab in the treatment of advanced colorectal cancer. Br J Cancer. 2014;110(3):648–55.
Qin S, Xu J, Wang L, Cheng Y, Liu T, Chen J, et al. Impact of tumor epidermal growth factor receptor (EGFR) status on the outcomes of first-line FOLFOX-4±cetuximab in patients (pts) with RAS-wild-type (wt) metastatic colorectal cancer (mCRC) in the randomized phase 3 TAILOR trial. Ann Oncol. 2016;27:vi174.
Qin S, Li J, Wang L, Xu J, Cheng Y, Bai Y, et al. Efficacy and Tolerability of First-Line Cetuximab Plus Leucovorin, Fluorouracil, and Oxaliplatin (FOLFOX-4) Versus FOLFOX-4 in Patients With RAS Wild-Type Metastatic Colorectal Cancer: the open-label, randomized, Phase III TAILOR Trial. J Clin Oncol. 2018;36(30):3031–9.
Seligmann J, Wood H, Richman S, Elliott F, Taylor M, Tinkler-Hundal E, et al. Epidermal growth factor receptor (EGFR) copy number (CN) as a biomarker of prognosis and panitumumab (Pan) benefit in RAS-wt advanced colorectal cancer (aCRC). Ann Oncol. 2017;28: v185.
Seligmann JF, Elliott F, Richman SD, Jacobs B, Hemmings G, Brown S, et al. Combined epiregulin and amphiregulin expression levels as a predictive biomarker for panitumumab therapy benefit or lack of benefit in patients with RAS wild-type advanced colorectal cancer. JAMA Oncol. 2016;2(5):633–42.
Williams CJM, Seligmann JF, Elliott F, Shires M, Richman SD, Brown S, et al. Artificial Intelligence-assisted amphiregulin and epiregulin IHC predicts panitumumab benefit in RAS wild-type metastatic colorectal cancer. Clin Cancer Res. 2021;27(12):3422–31.
Seymour MT, Brown SR, Middleton G, Maughan T, Richman S, Gwyther S, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14(8):749–59.
Seligmann JF, Hatch AJ, Richman SD, Elliott F, Jacobs B, Brown S, et al. Association of tumor HER3 messenger RNA expression with panitumumab efficacy in advanced colorectal cancer. JAMA Oncol. 2018;4(4):564–8.
Laurent-Puig P, Paget-Bailly S, Vernerey D, Vazart C, Decaulne V, Fontaine K, et al. Evaluation of miR 31 3p as a biomarker of prognosis and panitumumab benefit in RAS-wt advanced colorectal cancer (aCRC): Analysis of patients (pts) from the PICCOLO trial. J Clin Oncol. 2015;33(15).
Smith CG, Fisher D, Claes B, Maughan TS, Idziaszczyk S, Peuteman G, et al. Somatic profiling of the epidermal growth factor receptor pathway in tumors from patients with advanced colorectal cancer treated with chemotherapy ± cetuximab. Clin Cancer Res. 2013;19(15):4104–13.
Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. The Lancet. 2011;377(9783):2103–14.
Adams RA, Fisher D, Farragher S, Scott A, Smith CG, James MD, et al. Epiregulin (EREG) and amphiregulin (AREG) gene expression to predict response to cetuximab therapy in combination with oxaliplatin (Ox) and 5FU in first-line treatment of advanced colorectal cancer (aCRC). J Clin Oncol. 2012;30(15).
Guren TK, Thomsen M, Kure EH, Sorbye H, Glimelius B, Pfeiffer P, et al. Cetuximab in treatment of metastatic colorectal cancer: final survival analyses and extended RAS data from the NORDIC-VII study. Br J Cancer. 2017;116(10):1271–8.
Ciardiello F, Normanno N, Martinelli E, Troiani T, Pisconti S, Cardone C, et al. Cetuximab continuation after first progression in metastatic colorectal cancer (CAPRI-GOIM): A randomized phase II trial of FOLFOX plus cetuximab versus FOLFOX. Ann Oncol. 2016;27(6):1055–61.
Cushman SM, Jiang C, Hatch AJ, Shterev I, Sibley AB, Niedzwiecki D, et al. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: results from CALGB 80203 (Alliance). Clin Cancer Res. 2015;21(5):1078–86.
Laurent-Puig P, Grisoni ML, Heinemann V, Liebaert F, Neureiter D, Jung A, et al. Validation of miR-31-3p expression to predict cetuximab efficacy when used as first-line treatment in RAS wild-type metastatic colorectal cancer. Clin Cancer Res. 2019;25(1):134–41.
Miller-Phillips L, Heinemann V, Stahler A, Von Weikersthal LF, Kaiser F, Al-Batran SE, et al. Association of microRNA-21 with efficacy of cetuximab in RAS wild-type patients in the FIRE-3 study (AIO KRK-0306) and microRNA- 21's influence on gene expression in the EGFR signaling pathway. J Clin Oncol 2019;37.
Innocenti F, Ou FS, Qu X, Zemla TJ, Niedzwiecki D, Tam R, et al. Mutational analysis of patients with colorectal cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol. 2019;37(14):1217–27.
Rivera F, Karthaus M, Hecht JR, Sevilla I, Forget F, Fasola G, et al. Final analysis of the randomised PEAK trial: overall survival and tumour responses during first-line treatment with mFOLFOX6 plus either panitumumab or bevacizumab in patients with metastatic colorectal carcinoma. Int J Colorectal Dis. 2017;32(8):1179–90.
Laurent-Puig P, Bridgewater JA, Primrose JN, Pugh SA, Thomas G, Moutasim K, et al. Mir-31–3p as a predictive biomarker of cetuximab effects in a post hoc analysis of new EPOC phase III trial. J Clin Oncol. 2014;32(15).
Pugh S, Thiébaut R, Bridgewater J, Grisoni ML, Moutasim K, Rousseau F, et al. Association between miR-31-3p expression and cetuximab efficacy in patients with KRAS wild-type metastatic colorectal cancer: a post-hoc analysis of the New EPOC trial. Oncotarget. 2017;8(55):93856–66.
Sastre J, García-Alfonso P, Viéitez JM, Cano MT, Rivera F, Reina-Zoilo JJ, et al. Influence of BRAF and PIK3CA mutations on the efficacy of FOLFIRI plus bevacizumab or cetuximab as first-line therapy in patients with RAS wild-type metastatic colorectal carcinoma and <3 baseline circulating tumour cells: the randomised phase II VISNÚ-2 study. ESMO Open. 2021;6(2): 100062.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12(6):594–603.
Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28(7):1254–61.
Cui D, Cao D, Yang Y, Qiu M, Huang Y, Yi C. Effect of BRAF V600E mutation on tumor response of anti-EGFR monoclonal antibodies for first-line metastatic colorectal cancer treatment: a meta-analysis of randomized studies. Mol Biol Rep. 2014;41(3):1291–8.
Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51(5):587–94.
Stintzing S, Heinrich K, Tougeron D, Modest DP, Schwaner I, Euker J, et al. Randomized study to investigate FOLFOXIRI plus either bevacizumab or cetuximab as first-line treatment of BRAF V600E-mutant mCRC: The phase-II FIRE-4.5 study (AIO KRK-0116). Wolters Kluwer Health; 2021.
Wu S, Gan Y, Wang X, Liu J, Li M, Tang Y. PIK3CA mutation is associated with poor survival among patients with metastatic colorectal cancer following anti-EGFR monoclonal antibody therapy: a meta-analysis. J Cancer Res Clin Oncol. 2013;139(5):891–900.
Mao C, Yang ZY, Hu XF, Chen Q, Tang JL. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2012;23(6):1518–25.
Mohamed A, Twardy B, AbdAllah N, Akhras A, Ismail H, Zordok M, et al. Clinical impact of PI3K/BRAF Mutations in RAS wild metastatic colorectal cancer: meta-analysis results. J Gastrointest Cancer. 2019;50(2):269–75.
Wang ZH, Gao QY, Fang JY. Loss of PTEN expression as a predictor of resistance to anti-EGFR monoclonal therapy in metastatic colorectal cancer: evidence from retrospective studies. Cancer Chemother Pharmacol. 2012;69(6):1647–55.
Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014;53(7):852–64.
Li XD, Miao SY, Wang GL, Yang L, Shu YQ, Yin YM. Amphiregulin and epiregulin expression in colorectal carcinoma and the correlation with clinicopathological characteristics. Onkologie. 2010;33(7):353–8.
Dong J, Opresko LK, Dempsey PJ, Lauffenburger DA, Coffey RJ, Wiley HS. Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96(11):6235–40.
Oliveras-Ferraros C, Cufí S, Queralt B, Vazquez-Martin A, Martin-Castillo B, de Llorens R, et al. Cross-suppression of EGFR ligands amphiregulin and epiregulin and de-repression of FGFR3 signalling contribute to cetuximab resistance in wild-type KRAS tumour cells. Br J Cancer. 2012;106(8):1406–14.
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):3230–7.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–30.
Personeni N, Fieuws S, Piessevaux H, De Hertogh G, De Schutter J, Biesmans B, et al. Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin Cancer Res. 2008;14(18):5869–76.
Uhlyarik A, Piurko V, Papai Z, Raso E, Lahm E, Kiss E, et al. EGFR Protein Expression in KRAS Wild-Type Metastatic Colorectal Cancer Is Another Negative Predictive Factor of the Cetuximab Therapy. Cancers (Basel). 2020;12(3):614.
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27(30):5068–74.
Baker JB, Dutta D, Watson D, Maddala T, Munneke BM, Shak S, et al. Tumour gene expression predicts response to cetuximab in patients with KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2011;104(3):488–95.
Jing C, Jin YH, You Z, Qiong Q, Jun Z. Prognostic value of amphiregulin and epiregulin mRNA expression in metastatic colorectal cancer patients. Oncotarget. 2016;7(34):55890–9.
Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–25.
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100(15):8933–8.
Barber PR, Weitsman G, Lawler K, Barrett JE, Rowley M, Rodriguez-Justo M, et al. HER2-HER3 Heterodimer Quantification by FRET-FLIM and Patient Subclass Analysis of the COIN Colorectal Trial. J Natl Cancer Inst. 2020;112(9):944–54.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–23.
Finocchiaro G, Cappuzzo F, Janne P, Bencardino K, Carnaghi C, Franklin W, et al. EGFR, HER2 and Kras as predictive factors for cetuximab sensitivity in colorectal cancer. J Clin Oncol. 2007;25(18_suppl):4021.
Martin V, Landi L, Molinari F, Fountzilas G, Geva R, Riva A, et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br J Cancer. 2013;108(3):668–75.
Scartozzi M, Giampieri R, Maccaroni E, Mandolesi A, Giustini L, Silva R, et al. Analysis of HER-3, insulin growth factor-1, nuclear factor-kB and epidermal growth factor receptor gene copy number in the prediction of clinical outcome for K-RAS wild-type colorectal cancer patients receiving irinotecan-cetuximab. Ann Oncol. 2012;23(7):1706–12.
Scartozzi M, Mandolesi A, Giampieri R, Bittoni A, Pierantoni C, Zaniboni A, et al. The role of HER-3 expression in the prediction of clinical outcome for advanced colorectal cancer patients receiving irinotecan and cetuximab. Oncologist. 2011;16(1):53–60.
Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15(6):321–33.
Chen B, Xia Z, Deng YN, Yang Y, Zhang P, Zhu H, et al. Emerging microRNA biomarkers for colorectal cancer diagnosis and prognosis. Open Biol. 2019;9(1): 180212.
To KK, Tong CW, Wu M, Cho WC. MicroRNAs in the prognosis and therapy of colorectal cancer: from bench to bedside. World J Gastroenterol. 2018;24(27):2949–73.
Hollis M, Nair K, Vyas A, Chaturvedi LS, Gambhir S, Vyas D. MicroRNAs potential utility in colon cancer: Early detection, prognosis, and chemosensitivity. World J Gastroenterol. 2015;21(27):8284–92.
Wang CJ, Zhou ZG, Wang L, Yang L, Zhou B, Gu J, et al. Clinicopathological significance of microRNA-31, -143 and -145 expression in colorectal cancer. Dis Markers. 2009;26(1):27–34.
Sun D, Yu F, Ma Y, Zhao R, Chen X, Zhu J, et al. MicroRNA-31 activates the RAS pathway and functions as an oncogenic MicroRNA in human colorectal cancer by repressing RAS p21 GTPase activating protein 1 (RASA1). J Biol Chem. 2013;288(13):9508–18.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (No.82222074, 81874436, 82074154, 81774240), Three-year Action Plan for the Development of Chinese Medicine in Shanghai (No. ZY (2018–2020)-CCCX-2003–01), Shanghai Key Clinical Specialty Construction Project (No.shslczdzk01201), The Siming Scholar from Shanghai Shuguang Hospital (SGXZ-201904).
Author information
Authors and Affiliations
Contributions
Z.Y. and Y.G. conceived the study. X.L., Y.L. designed research methods. Y.L., X.Z., and J.S. did the literature search and collected the data. H.F. and Z.Y. checked the data. X.L. and Y.L. performed the statistical analyses and wrote the manuscript. H.F. and Y.G. proofread the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Appendix A. Detailed search strategy
Additional file 2: Supplementary Table S1.
PRISMA checklist.
Additional file 3:
Supplementary Table S2. 30 publications included in the review.
Additional file 4:
Supplementary Table S3. Risk of Bias Assessment using the Newcastle-Ottawa Scalea.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, X., Li, Y., Li, Y. et al. Prognostic and predictive biomarkers for anti-EGFR monoclonal antibody therapy in RAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. BMC Cancer 23, 1117 (2023). https://doi.org/10.1186/s12885-023-11600-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-11600-z