
Abstract
Background
Dysbiosis of gut microbiota has been linked to numerous diseases, including cancer. The unique role of gut microbiota in urological tumors is gaining prominence. However, it is still controversial whether the dysbiosis of gut microbiota should be one of the etiological factors of bladder cancer (BCa), prostate cancer (PCa) or kidney cancer (KCa).
Materials and methods
The microbiome genome-wide association study (GWAS) from the MiBioGen consortium (18,340 samples of 24 population-based cohorts) was utilized as the exposure data. Additionally, outcomes data (951 BCa cases and 307,092 controls; 1,631 KCa cases and 238,678 controls; 79,148 PCa cases and 61,106 controls) were extracted from the GWAS of the FinnGen and PRACTICAL consortia. To detect the potential causative bacterial traits for BCa, PCa, and KCa, a two-sample Mendelian randomization (MR) analysis was performed, employing the inverse-variance weighted or Wald ratio method. Sensitivity analyses were subsequently conducted to explore the robustness of the primary results. Finally, the reverse MR analysis was undertaken to mitigate the reverse causation.
Results
This study suggested that Bifidobacterium (p = 0.030), Actinobacteria (p = 0.037 for phylum, 0.041 for class), and Ruminococcustorques group (p = 0.018), exhibited an association with an increased risk of BCa using either the inverse-variance weighted or Wald ratio method. By utilizing the Wald ratio method, Allisonella (p = 0.004, p = 0.038) was associated with a decreased risk of BCa and PCa, respectively. Furthermore, Ruminococcustorques group (p = 0.028) and Erysipelatoclostridium (p = 0.048) were causally linked to an elevated risk of KCa.
Conclusions
This MR study supports that genetically predicted gut microbiota is causally related to BCa, PCa and KCa. Additionally, distinct bacterial traits are identified in relation to each tumor type.
Similar content being viewed by others


Introduction
Urological tumors pose a severe threat to human life and health on a global scale. According to the Global Burden of Disease Study (2019), the incidence rates of bladder cancer (BCa), prostate cancer (PCa), and kidney cancer (KCa) stand at 6.5, 17.4, and 4.6 per 100,000 person-years, collectively accounting for 9.81% of the worldwide tumor incidence. The mortality rates of PCa, BCa, and KCa are respectively 6.3, 2.9, and 2.1 per 100,000 person-years, contributing to approximately 9.14% of all tumor-related fatalities [1]. Furthermore, variations in incidence rates are observed globally, with higher incidence rates in industrialized nations like the United States and Europe. Meanwhile, the incidence rates of South American countries are higher than rising nations like African and Asian [2,3,4]. In recent decades, with the application of immunotherapy in tumors, focusing on the organism immunity of tumors and the various factors that regulate the immune status is increasing, and gut microbiota is one of them [5]. Since the Human Microbiome Project launched, there has been increasing evidence to suggest that the microbiome, including urinary tract, gut, and intratumoral microbes, contributes to urologic tumorigenesis [6].
Recently, gut microbiota has gained widespread attention as a remarkable factor to regulate the health of organism. Dysbiosis of gut microbiota can regulate immune, energy, lipid and glucose metabolism pathways that involved in the development of diseases, such as obesity, type 2 diabetes, hepatic steatosis, and cancer [7]. The gut microbiota plays an important role in the development of various gastrointestinal disorders, including colorectal cancer and inflammatory bowel disease, for which digestive tract is habitat of gut microbiota [8]. With organs indirect contact with each other, it remains unclear whether a relevancy exists between gut microbiota and urinary system. According to some preliminary investigations, gut microbiota affects the growth of PCa, BCa, and KCa through their metabolites [9,10,11]. Matsushita M et al. reported that short-chain fatty acids (SCFAs) producing bacteria, namely Rikenellaceae, Alistipes, and Lachnospira, of those which were considerably increased in men with high Gleason prostate cancer [12]. He et al. demonstrated that among BCa patients, Prevotella, Clostridium cluster XI, and the concentration of butyric acid in feces were significantly reduced [13]. Dai et al. reported that the disturbance of tryptophan metabolites in gut microbiota is associated with renal cancer metastasis [11]. However, due to the scarcity of large-scale real-world studies, these observations remain to be definitively substantiated. Furthermore, investigations into gut microbiota are inherently challenging and resource-intensive, requiring advanced molecular tools and techniques (i.e., macrogenome, metabolome, lipidome, and macrotranscriptome) while grappling with the complexities of confounding factors, biases, and reverse causation in general observational studies.
Genome-wide association studies (GWAS) detect genetic variants across large populations to verify phenotype-genotype associations, and more than 50,000 associations of genome-wide significance have been reported in various common diseases and traits [14]. MR is a statistical method that leverages GWAS data as distinct phenotypes to address the limitations of observational studies. By considering genetic variants as instrumental variables (IVs), MR seeks to uncover the potential causal links between exposures and outcomes [15]. MR has been widely applied in research on causal inference. Recently, MR investigations have revealed the causality between gut microbiota and several illnesses such as colorectal cancer, Alzheimer’s disease, autoimmune diseases, or psychiatric disorders [16,17,18,19].
This study aims to investigate the potential causality between gut microbiota and urological tumors, specifically prostate cancer, bladder cancer, and renal cancer, by employing the two-sample MR method.
Materials and methods
Study design overview
This study employs the two-sample MR method to investigate the causal associations between gut microbiota and PCa, BCa, and/or KCa. Summary statistic data for gut microbiota and PCa, BCa, or KCa were extracted from the substantial GWASs to select the IVs. Under the condition of published research and open-access summary data were used, thus further ethical approval or participant consent was unnecessary.
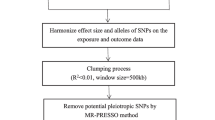
To mitigate the potential biases on the results, adherence to three major assumptions in MR method is crucial: (1) IVs must exhibit a significant association with the exposure [20]; (2) IVs should exclusively influence the outcome via the exposure [20]; and (3) IVs must not be linked to the outcome due to confounding factors [20]. An overview of the study design is illustrated in Fig. 1.

The overview of design. IVs: instrumental variables; GWAS: Genome Wide Association Study; LD: linkage disequilibrium; SNP: single nucleotide polymorphism; BCa: bladder cancer; PCa: prostate cancer; KCa: kidney cancer
Date sources
Gut microbiota
A large-scale GWAS involved 18,340 participants from 24 cohorts provides summary statistics for gut microbiota, using 16 S rRNA gene sequencing [21]. Available data were from the MiBioGen (https://mibiogen.gcc.rug.nl/). In all, 211 traits (131 genera, 35 families, 20 orders, 16 classes, and 9 phyla) were included [21]. First, IVs were selected at p < 5 × 10−8 to meet the stricter threshold. Subsequently, linkage disequilibrium (LD) clumping was executed to mitigate LD among single nucleotide polymorphisms (SNPs) (r2 < 0.001, distance = 10,000 kb), resulting in an adjusted cutoff (r2 < 0.01, distance = 500 kb) to retain a viable number of SNPs for analysis. SNPs without attribution to specific bacterial traits were excluded. Finally, “PhenoScanner” (http://www.phenoscanner.medschl.cam.ac.uk/) was used to exclude SNPs that were clearly associated with risk factors of the urologic tumors. A total of 1,393 SNPs closely related to 25 bacterial traits were incorporated into the MR analysis.
PCa, BCa, and KCa
Summary statistics for PCa (79,148 cases and 61,106 controls of European descent) were furnished by the Prostate Cancer Association Group to Investigate Cancer-Associated Alterations in the Genome (PRACTICAL, http://practical.icr.ac.uk) consortium [22]. FinnGen research project (https://www.finngen.fi), which involves participates of European descent, provided summary statistics for BCa (2,072 cases, 238,678 controls,) and KCa (1,631 cases, 238,678 controls) [23].
Statistical analysis
F-statistics were employed to test the strength of IVs, mitigating potential weak instrument bias which could confound causal association estimates. F-statistics were calculated through the following formula: F = R2 (n-k-1)/k(1-R2), where “n” signifies the sample size, “k” denotes the number of IVs and “R2” represents the portion of exposure variance elucidated by the IVs [24]. Generally, R2 was estimated via employing the equation required minor allele frequency (MAF) and β value: R2 = 2 × MAF × (1 − MAF) × β2. Due to the lack of MAF value in GWAS, the function “get_r_from_bsen” of “TwoSampleMR” package was employed for R2 estimation. The weak IVs were discarded with F-statistics < 10.
In this study, the MR analysis involving bacterial traits linked to individual SNPs was conducted using the Wald ratio method. Multiple tests, encompassing the inverse variance weighted (IVW) method, weighted median method, and MR-Egger regression test, were conducted for bacterial traits with multiple associated SNPs [25]. The results of the IVW method were plausible if the three assumptions of MR were satisfied for SNPs [25]. The Cochrane’s Q test was performed to scrutinize SNP-associated heterogeneity for each bacterial trait. In cases where significant heterogeneity emerged (p < 0.05), a fixed-effects IVW model was applied; contrarily, a random-effects IVW model was applied [26]. Additionally, MR-Egger intercept test and leave-one-out analysis were performed for sensitivity analysis [27]. The p-value of MR-Egger intercept test functioned as an indicator of the horizontal pleiotropy (statistically significant if p < 0.05) [28]. Leave-one-out analysis was used to discern potential pleiotropic effects stemming from individual SNP. Finally, reverse MR analysis was executed to verify the existence of reverse causality between BCa, PCa, KCa, and gut microbiota. All MR analyses were conducted utilizing “TwoSampleMR” R package (version 4.2.1).
Results
Overview of instrumental variables
Following a sequence of rigorous quality control procedures, 27 SNP (p < 5 × 10−8, R2 < 0.01) associated with 19 bacterial traits were selected as IVs for analysis (Supplementary Table 1). The IVs employed in the MR analysis possessed F-statistics within a range of 30.07 to 200.70, all of which exceeded the threshold of > 10. This indicates a robust instrument strength and mitigates the potential impact of weak instrument bias (Supplementary Table 2). Limited by the number of available IVs, the sensitivity analysis was restricted to the Bifidobacterium. Notably, the statistical effect size remained relatively consistent across taxonomic levels, encompassing order, family, and genus.
Gut microbiota and BCa
In the context of MR analysis, a comprehensive assessment revealed that 7 bacterial traits (encompassing various taxonomic levels of phylum, class, order, family, and genus) exhibited statistically associations with the risk of BCa. This suggests a potential role for specific bacterial traits in the etiology of BCa (Table 1; Fig. 2).

The forest plots of major results. SNP: single nucleotide polymorphism; BCa: bladder cancer; PCa: prostate cancer; KCa: kidney cancer; IVW: inverse-variance weighted; OR: odds ratio
Among the aforementioned traits, Bifidobacterium, Bifidobacteriaceae, Bifidobacteriales were found to be the same category of bacteria, sharing identical IVs represented by rs182549, rs7322849, and rs7570971. As demonstrated in Table 1, the IVW analysis was concluded that Bifidobacterium exhibited a causal relationship with an elevated risk of BCa (OR: 1.496, 95% CI: 1.039–2.154, p = 0.030); similar results were obtained for Bifidobacterium and Bifidobacteriaceae (OR: 1.505, 95% CI: 1.040–2.179, p = 0.030; OR: 1.505, 95% CI: 1.040–2.180, P = 0.030, respectively). The results also remained robust in weighted median analyses (OR: 1.512, 95% CI: 1.011–2.260, p = 0.044; OR: 1.522, 95% CI: 1.005–2.305, p = 0.047; OR: 1.522, 95% CI: 1.012–2.290, p = 0.043, respectively). The causal relationship observed within the higher taxonomic level of Bifidobacteria, specifically Actinobacteria (phylum) and Actinobacteria (class), echoed the findings with an increased risk of BCa (OR: 1.765, 95% CI: 1.034–3.013, p = 0.037; OR: 1.546: 95% CI: 1.018–2.349, p = 0.041, respectively). Additionally, Ruminococcustorques group and Allisonella, each had only one SNP, exhibited potential causal relationships as determined by Wald ratio analyses (OR: 3.656, 95% CI: 1.248–10.706, p = 0.018; OR: 0.534, 95% CI: 0.348–0.818, p = 0.004, respectively).
In the subsequent analyses, Cochran’s Q test did not reveal any statistically significant heterogeneity for any of the applicable bacterial traits (p > 0.10, Supplementary Table 3). A leave-one-out analysis was applied and identified no single SNP with a significant influence on the IVW estimate (Supplementary Fig. 1). Furthermore, less directional pleiotropy was inspected in the MR-Egger test (Bifidobacterium, intercept p = 0.964; Bifidobacteriaceae, intercept p = 0.944; Bifidobacteriales, intercept p = 0.944, Supplementary Table 4). Considering the absence of enough SNPs, a sensitivity analysis is not feasible for other bacterial traits.
Gut microbiota and PCa or KCa
Genetical evidence indicated a negative effect of Allisonella on the risk of PCa (OR: 0.894, 95%CI: 0.805–0.994, p = 0.038) as determined through Wald ratio analysis (Table 1; Fig. 2). In parallel, Ruminococcustorques group and Erysipelatoclostridium were causally associated with an elevated risk of KCa (OR: 3.798, 95% CI: 1.154–12.504, p = 0.028; OR: 2.310, 95% CI: 1.007–5.301, p = 0.048, respectively) as indicated by Wald ratio analysis (Table 1; Fig. 2). There were not enough SNPs to conduct a sensitivity analysis for the aforementioned bacterial traits.
Reverse MR analysis
Adopting a consistent threshold with the main analysis for SNP selection, reverse MR analysis failed to identify any causal relationships between BCa, PCa, KCa, and gut microbiota (Supplementary Tables 5, 6).
Discussion
This study pioneers the utilization of a two-sample MR approach to identify a potential causal relationship between specific gut microbiota taxa and three major urological tumors: BCa, PCa, and KCa, which provides directions for further mechanistic investigations.
The human microbiota resides ubiquitously across the body surface and natural cavities, forming a harmonious symbiotic equilibrium [1]. Therefore, the intricate interplay between human microbiota and organismal health has long been a focal point of research. Numerous existing researches have revealed the potential influence of urinary system bacteria on urological tumor development, the widespread concern in the unique role of gut microbiota in neoplastic diseases has sparked novel inquiries into the relationship between gut microbiota and urological tumors [29, 30].
This two-sample MR analysis unveiled a surprising increase in the risk of BCa associated with Bifidobacterium at the order, family, and genus levels, Actinobacteria at the phylum and class levels, Ruminococcustorques group at the genus level, while Allisonella at the genus level appeared to confer a potential protective effect against BCa. Bifidobacterium has long been recognized as a probiotic abundant in fermented dairy products. Studies have indicated that the intake of fermented dairy foods is linked to a reduced risk of BCa [31]. Bifidobacterium may play a role in the regulation of proliferation, apoptosis, responses to immune therapy, radiation, and chemotherapy [32]. However, existing research on the anticancer benefits of probiotics are primarily concentrated on the intestinal tract tumors, with a lot of unknown of their impact on tumors in other organs [32]. Given that the interactions in the intestinal tract are more direct and intricate due to the site’s flora aggregation, it remains questionable whether the preliminary findings regarding Bifidobacterium, or even probiotics in general, and their effects on intestinal tumors can be extrapolated to tumors in other organs. Moreover, Lactobacillus, including Bifidobacterium, has been implicated in promoting the pathogenesis of gastric cancer through diverse mechanisms, including supplying exogenous lactic acid, stimulating inflammation, angiogenesis, and epithelial-mesenchymal transition [33]. Thus, this study has a proposal in a novel avenue of etiological evidence indicating that Bifidobacterium could potentially contribute to BCa development. At present, a dearth of research exists regarding the specific mechanism underlying the relationship between Bifidobacterium and BCa. Future exploration is essential to determine whether intestinal Bifidobacterium influences critical physiological activities of urothelial cells through various small molecular metabolites or via translocation and colonization of the urinary tract. Speculatively, based on current knowledge, Bifidobacterium may possess the ability to stimulate macrophages, T lymphocytes, and epithelial cells to secrete tumor necrosis factor α, which in turn could promote tumor proliferation, survival, and evasion from immune surveillance—this represents a potentially promising mechanistic pathway [34, 35]. In terms of observational studies, a preliminary study of gut microbiota in BCa patients, comprised 26 cases and 16 health controls, reported decreased gut microbial diversity at the phylum level, with decreased relative quantities of Clostridium cluster XI and Prevotella in BCa patients; however, no significant difference was observed in relation to Bifidobacterium [13]. Emerging evidence suggests that gut microbiota could influence BCa treatment outcomes. Non-muscle invasive BCa patients treated with probiotics exhibited lower recurrence rates [36]. Recently, the first fecal macro-genomic research of BCa was published, encompassing 32 cases and 15 health controls, confirmed the viewpoint of gut microbiota dysbiosis in BCa and revealed changes in key metabolites. Furthermore, the relative abundance of 19 microbiota at the genus level (including Bifidobacterium) was diminished in fecal samples from BCa patients [37]. Besides the unclear reverse causality and limited sample size, the inclusion and exclusion criteria which only excluded vegetarians, failed to account for the potential confounding introduced by various dietary habits, and the data of vegetarians were omitted. Consequently, the identification of relevant discrepancies necessitates further investigation.
This study also identified that at the genus level, Allisonella decreased the risk of PCa. The intricate interplay between gut microbiota and PCa has recently garnered significant attention, leading to the emergence of “gut-prostate axis” concept. However, the precise mechanisms remain elusive. Serveral observational studies have individually suggested potential associations between PCa and various bacteria, including Streptococcus, Bacteroides, massiliensis, Prevotella 9, Erysipelotrichaceae, Escherichia/Shigella, Rikenellaceae, Alistipes or Lachnospira [12, 38,39,40]. This study uniquely highlighted the role of Allisonella while meticulously addressing reverse causality and confounding issues. Metabolites produced by gut microbiota, such as SCFAs, may regulate PCa growth, and more significantly, androgen production by gut microbiota could contribute to the development of castration-resistant prostate cancer [9]. Due to the intricate nature of gut microbiota, causality should be interpreted cautiously; nonetheless, the undeniable significance of gut microbiota in the evolution of prostate cancer is evident.
Ruminococcustorques group and Erysipelatoclostridium at genus level were identified as potential contributors to KCa occurrence. The sole available observational study (comprising 50 cases and 40 health controls) reported positive associations between Blautia, Streptococcus, Ruminococcustorques_group, Romboutsia, and Eubacteriumhallii group with renal clear cell cancer. This study also indicated that Streptococcus promotes renal clear cell cancer progression in vitro through the TGF-β signaling pathway [41]. While consensus exists regarding the potential involvement of the Ruminococcustorques group in the occurrence of KCa, the substantiation of this correlation necessitates more robust empirical evidence.
Several limitations are existed in this study. Firstly, the findings are limited to European lineages, thus the substantial variations in gut microbiota composition across different populations were not considered. Secondly, the 16 S rRNA gene sequencing method only allowed discrimination from the phylum level to the genus level and not at a more specific taxonomic level. Thirdly, it is unfit to conduct stratified MR due to the unavailability of population basic characteristics, such as gender, culture, occupation, etc., which bring about a lack of explanatory power for the hazards associated with particular populations. Finally, gut microbiota may be influenced by dietary habits or other environmental factors, yet they were unable to assess whether genetic instrumental variables were correlated with these confounding factors due to the unavailability of relevant information.
Conclusions
This MR study supports genetic evidence that gut microbiota is causally related to BCa, PCa or KCa, and specific bacterial traits are suggested separately. The results offer a fresh perspective for exploring the intricate mechanisms through which gut microbiota affects urological tumors.
Availability of data and materials
The original statistical data analyzed in the study are presented in the article, which is open access.
Abbreviations
- MR:
-
Mendelian randomization
- BCA:
-
Bladder cancer
- PCA:
-
Prostate cancer
- KCA:
-
Kidney cancer
- GWAS:
-
Genome-wide association
- IVs:
-
Instrumental variables
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- SNP:
-
Single nucleotide polymorphism
- IVW:
-
Inverse variance weighted
- SCFAs:
-
Short-chain fatty acids
References
Kocarnik JM, Compton K, Dean FE, Fu W, Gaw BL, Harvey JD, et al. Cancer Incidence, Mortality, Years of Life Lost, Years lived with disability, and disability-adjusted life years for 29 Cancer Groups from 2010 to 2019: a systematic analysis for the global burden of Disease Study 2019. JAMA Oncol. 2022;8(3):420–44.
Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE, et al. Prostate cancer. Nat Rev Dis Primers. 2021;7(1):9.
Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder Cancer incidence and mortality: A global overview and recent Trends. Eur Urol. 2017;71(1):96–108.
Capitanio U, Bensalah K, Bex A, Boorjian SA, Bray F, Coleman J, et al. Epidemiology of renal cell carcinoma. Eur Urol. 2019;75(1):74–84.
Garrett WS. Cancer and the microbiota. Science. 2015;348(6230):80–6.
Kustrimovic N, Bombelli R, Baci D, Mortara L. Microbiome and prostate Cancer: a Novel Target for Prevention and Treatment. Int J Mol Sci. 2023;24(2);1511.
de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. 2022;71(5):1020–32.
Marchesi JR, Adams DH, Fava F, Hermes GD, Hirschfield GM, Hold G, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65(2):330–9.
Fujita K, Matsushita M, Banno E, De Velasco MA, Hatano K, Nonomura N, et al. Gut microbiome and prostate cancer. Int J Urol. 2022;29(8):793–8.
Martin A, Woolbright BL, Umar S, Ingersoll MA, Taylor JA. 3rd. Bladder cancer, inflammageing and microbiomes. Nat Rev Urol. 2022;19(8):495–509.
Dai G, Chen X, He Y. The gut microbiota activates AhR through the Tryptophan Metabolite Kyn to Mediate Renal Cell Carcinoma Metastasis. Front Nutr. 2021;8:712327.
Matsushita M, Fujita K, Motooka D, Hatano K, Fukae S, Kawamura N, et al. The gut microbiota associated with high-gleason prostate cancer. Cancer Sci. 2021;112(8):3125–35.
He C, Li B, Huang L, Teng C, Bao Y, Ren M, et al. Gut microbial composition changes in bladder cancer patients: a case-control study in Harbin, China. Asia Pac J Clin Nutr. 2020;29(2):395–403.
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20(8):467–84.
Emdin CA, Khera AV, Kathiresan S. Mendelian Randomization Jama. 2017;318(19):1925–6.
Xu Q, Ni JJ, Han BX, Yan SS, Wei XT, Feng GJ, et al. Causal relationship between Gut Microbiota and Autoimmune Diseases: a two-sample mendelian randomization study. Front Immunol. 2021;12:746998.
Ni JJ, Xu Q, Yan SS, Han BX, Zhang H, Wei XT, et al. Gut Microbiota and Psychiatric Disorders: a two-sample mendelian randomization study. Front Microbiol. 2021;12:737197.
Zhuang Z, Yang R, Wang W, Qi L, Huang T. Associations between gut microbiota and Alzheimer’s disease, major depressive disorder, and schizophrenia. J Neuroinflammation. 2020;17(1):288.
Ni JJ, Li XS, Zhang H, Xu Q, Wei XT, Feng GJ, et al. Mendelian randomization study of causal link from gut microbiota to colorectal cancer. BMC Cancer. 2022;22(1):1371.
Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing mendelian randomization investigations. Wellcome Open Res. 2019;4:186.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65.
Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50(7):928–36.
Kurki MI, Karjalainen J, Palta P, Sipilä TP, Kristiansson K, Donner KM, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–18. https://www.finngen.fi/en/access_results.
Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40(3):740–52.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65.
Greco MF, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34(21):2926–40.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some Invalid Instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Markowski MC, Boorjian SA, Burton JP, Hahn NM, Ingersoll MA, Maleki Vareki S, et al. The Microbiome and Genitourinary Cancer: a collaborative review. Eur Urol. 2019;75(4):637–46.
Park EM, Chelvanambi M, Bhutiani N, Kroemer G, Zitvogel L, Wargo JA. Targeting the gut and tumor microbiota in cancer. Nat Med. 2022;28(4):690–703.
Zhang K, Dai H, Liang W, Zhang L, Deng Z. Fermented dairy foods intake and risk of cancer. Int J Cancer. 2019;144(9):2099–108.
Badgeley A, Anwar H, Modi K, Murphy P, Lakshmikuttyamma A. Effect of probiotics and gut microbiota on anti-cancer drugs: mechanistic perspectives. Biochim Biophys Acta Rev Cancer. 2021;1875(1):188494.
Vinasco K, Mitchell HM, Kaakoush NO, Castaño-Rodríguez N. Microbial carcinogenesis: lactic acid bacteria in gastric cancer. Biochim Biophys Acta Rev Cancer. 2019;1872(2):188309.
Chen AY, Wolchok JD, Bass AR. TNF in the era of immune checkpoint inhibitors: friend or foe? Nat Rev Rheumatol. 2021;17(4):213–23.
Lim HJ, Shin HS. Antimicrobial and Immunomodulatory Effects of Bifidobacterium strains: a review. J Microbiol Biotechnol. 2020;30(12):1793–800.
Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström PU, Choi W, et al. Bladder cancer Lancet. 2016;388(10061):2796–810.
Qin C, Chen Z, Cao R, Shi M, Tian Y. Integrated Analysis of the fecal metagenome and metabolome in bladder Cancer in a Chinese Population. Genes (Basel). 2022;13(11);1967.
Liss MA, White JR, Goros M, Gelfond J, Leach R, Johnson-Pais T, et al. Metabolic biosynthesis pathways identified from fecal Microbiome Associated with prostate Cancer. Eur Urol. 2018;74(5):575–82.
Golombos DM, Ayangbesan A, O’Malley P, Lewicki P, Barlow L, Barbieri CE, et al. The role of gut microbiome in the pathogenesis of prostate Cancer: a prospective, Pilot Study. Urology. 2018;111:122–8.
Gut microbiota differs. Significantly between men with and without prostate cancer. Cancer. 2023;129(2):169–70.
Chen Y, Ma J, Dong Y, Yang Z, Zhao N, Liu Q, et al. Characteristics of gut microbiota in patients with Clear Cell Renal Cell Carcinoma. Front Microbiol. 2022;13:913718.
Acknowledgements
We are grateful to the MiBioGen study for opening source of the gut microbiota GWAS summary statistics, and the PRACTICAL study and FinnGen study for releasing the GWAS summary statistics of PCA, BCA and KCA.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82072833, 82272864).
Author information
Authors and Affiliations
Contributions
WMD, and GX designed the study. WMD, QYJ, and WMS searched and analyzed the data. WMD and GX drafted the article. and PH supervised the study. All authors approved the submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No further informed permission was needed for this study because it only used data that was already available to the public. All studies have received prior approval from the appropriate institutional review boards.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Detailed information of instrumental variables used in MR analyses. Supplementary Table 2. MR analysis of all gut microbiome. Supplementary Table 3. The heterogeneity results from the Cochran's Q test. Supplementary Table 4. Directional pleiotropy results from Egger intercept analysis. Supplementary Table 5. Detailed information of instrumental variables used in reverse MR analyses. Supplementary Table 6. Reverse MR analysis of all gut microbiome
Additional file 2: Supplementary Figure 1.
Leave-one-out analysis results of Bifidobacteriales(order),Bifidobacteriaceae(family), and, Bifidobacterium(genus)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mingdong, W., Xiang, G., Yongjun, Q. et al. Causal associations between gut microbiota and urological tumors: a two-sample mendelian randomization study. BMC Cancer 23, 854 (2023). https://doi.org/10.1186/s12885-023-11383-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-11383-3