
Abstract
Background
Precision oncology, defined as treatment of patients with targeted therapies matched to specific molecular alterations, has entered routine clinical practice. Particularly in patients with advanced cancer or hematologic malignancies, for whom no further standard therapies are available, this approach is increasingly applied as last resort option outside of the approved indication. However, data on patient outcomes are not systematically collected, analyzed, reported, and shared. We have initiated the INFINITY registry to provide evidence from routine clinical practice to fill this knowledge gap.
Methods
INFINITY is a retrospective, non-interventional cohort study conducted at approximately 100 sites in Germany (office-based oncologists/hematologists and hospitals). We aim to include 500 patients with advanced solid tumors or hematologic malignancies who received a non-standard targeted therapy based on potentially actionable molecular alterations or biomarkers. INFINITY aims to provide insights into the use of precision oncology in routine clinical practice within Germany. We systematically collect details on patient and disease characteristics, molecular testing, clinical decision-making, treatment, and outcome.
Discussion
INFINITY will provide evidence on the current biomarker landscape driving treatment decisions in routine clinical care. It will also provide insights on effectiveness of precision oncology approaches in general, and of specific drug class/alteration matches used outside their approved indications.
Trial registration
The study is registered at ClinicalTrials.gov, NCT04389541.
Similar content being viewed by others

Background
Oncology is currently experiencing a paradigm shift from a “one-drug-fits-all” treatment model towards a tailored, molecular targeted or biomarker-driven treatment approach [1,2,3,4]. The goal is to choose the most effective, best tolerated drug for the patient, based on the molecular profile of the individual disease.
To investigate the efficacy of this novel treatment approach, new clinical trial concepts have evolved, shifting from a tumor type focus to a molecular profile focus. Thus, biomarker-guided trial designs have recently emerged, such as basket trials allocating patients to matched treatments based on molecular alteration across cancer types, or umbrella trials involving one cancer type and different treatments matched to genomic alterations [2, 5].
Several precision oncology trials have shown promising results, suggesting that molecular-matched targeted therapy has the potential to improve outcomes compared to conventional therapy [2, 6,7,8,9,10,11]. However, it is difficult to conduct biomarker-driven clinical trials and thus, reliable evidence from randomized trials is still lacking. In this context, real-world data are a valuable data source with the potential to address and couple real-world molecular and clinical data and to provide deep insights also into sequential treatment decisions and overall outcomes.
The number of available targeted drugs is steadily increasing [12, 13]. First drugs have been approved in a tumor-agnostic manner based on the presence of molecular alterations instead of specific tumor types: In 2017, the Food and Drug Administration (FDA) granted the first tumor-agnostic approval for pembrolizumab for patients with unresectable or metastatic, microsatellite instability-high or mismatch repair deficient (MSI-H/dMMR) solid tumors. As of 2021, four tumor-agnostic FDA approvals have been granted for three drugs (pembrolizumab, larotrectinib and entrectinib) based on three molecular alterations (MSI-H/dMMR, neurotrophic tyrosine receptor kinase (NTRK) gene fusion and high tumor mutational burden (TMB-H)) [14]. Furthermore, a growing number of targeted therapies has been approved in more than one tumor type based on specific molecular alterations (e.g., BRAF, HER2/ERBB2, RET, BRCA1/2, PD-L1) including the approval of dabrafenib/trametinib for pediatric patients with BRAF V600E mutant glioma and selpercatinib, a selective kinase inhibitor for patients with rearranged during transfection (RET) fusion-positive tumor by 2023 [5, 15, 16].
Besides conventional diagnostic techniques including polymerase chain reaction (PCR), immunohistochemistry (IHC), (fluorescent) in situ hybridization ((F)ISH), reverse transcription PCR (RT-PCR) and microarrays, next generation sequencing (NGS) techniques have transformed the diagnostic field and are now broadly available [5, 11].
Increasing availability and use of NGS testing as well as targeted therapies nourish the hope that these new technologies will quickly translate into patient clinical benefits, especially in situations where no further standard treatment is available. However, clinical outcomes of this approach are not systematically collected, analyzed, and reported.
The currently available evidence on the benefit of molecular-matched therapies used outside of their approved indication still carries substantial uncertainties. The only randomized trial in the field (SHIVA) found no clear benefit for patients receiving molecular-matched treatments used outside their labeled indications compared to standard of care (PFS 2.3 vs. 2.0 months) [17]. However, this trial was conducted several years ago, and diagnostic techniques and availability of targeted therapies have improved since then. MyPathway, a multicenter, multiple basket phase IIa study, evaluates efficacy of treatments targeting molecular alterations of HER2, epidermal growth factor receptor (EGFR), BRAF and the Hedgehog pathway in subprotocols [18]. The TAPUR phase II multi-basket trial, investigating safety and efficacy of commercially available targeted antineoplastic drugs in patients with metastatic solid tumors harboring potentially actionable molecular alterations, was initiated in 2016 [19]. While proof for a clinical benefit is missing in some patient cohorts, encouraging results in other cohorts [20] provide evidence of the feasibility and potential value of the basket study design in precision medicine [18, 21, 22]. The TAPUR trial is still ongoing, but first study results already helped to further shape the field: Treatment of metastatic colorectal cancer patients with FMS-like tyrosine kinase-3 (FLT-3) amplification with sunitinib was found to be ineffective; while on the other hand pembrolizumab monotherapy was reported to have anti-tumor activity in metastatic breast cancer patients with TMB-H, supporting the recent FDA approval [21, 22].
The INFINITY registry will provide systematically collected real-world data on the use of molecular-matched targeted therapies outside of their approved indication for treatment of patients with advanced solid tumors or hematologic malignancies, for whom no standard therapy is available. INFINITY explores how precision oncology is applied in routine clinical practice in Germany, how decisions are made by the treating physicians, which diagnostic tests are utilized to identify potentially actionable alterations, which substance classes of targeted therapies are used, and which molecular alterations are targeted. Effectiveness of matched therapies in the total patient cohort and in patient subgroups will be analyzed, aiming to generate hypotheses on effective or ineffective drug class/alteration matches. Furthermore, a decentral tissue sample biobank is set up, collecting sample information for genomic analysis for future translational research projects.
Methods/design
Objectives
INFINITY collects real-world data on precision oncology in Germany with the objective to assess treatment and outcomes of adult patients with advanced solid tumors or hematologic malignancies who received a non-standard targeted therapy (NSTT; i.e., a targeted therapy used outside the labeled indication and not recommended in guidelines) based on an actionable molecular alteration or associated with biomarker(s). NSTT administered in this study will be matched to targetable genomic or proteomic alterations or matched to biomarkers (i.e. biomarker-driven precision immunotherapy / checkpoint inhibitor / targeted therapy based on biomarker(s) associated with treatment benefit also including (but not restricted to) upstream and/or downstream intracellular targets). In particular, data on patient and disease characteristics, molecular testing, physician´s decision making, treatment (assessed as substance class), and outcome will be evaluated.
Study design
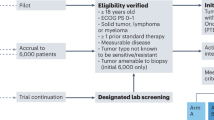
INFINITY is a retrospective, multicenter, non-interventional study, conducted at approximately 100 study sites in Germany, including hospitals and office-based oncologists. 500 patients will be enrolled over a recruitment period of 3 years, which started in April 2020.
Eligibility criteria
Inclusion criteria:
-
Patients with advanced solid tumors (i.e., locally advanced, inoperable and/or metastatic) or hematologic malignancies not eligible for standard therapy options (i.e., without further treatment options with drugs approved for the specific indication based on the judgement of the treating physician)
-
Started or completed a non-standard targeted therapy based on a potentially actionable alteration or biomarker identified by molecular diagnostics
-
Availability of molecular diagnostic test results that informed clinical decision to apply the non-standard targeted therapy
-
Targeted therapy must be non-standard at time point of patient enrollment/registration
-
Age ≥ 18 years
-
Signed and dated informed consent form (only if patient is alive at time of data entry into the project, not applicable for inclusion of deceased patients’ data)
Exclusion criteria:
-
The non-standard targeted therapy was administered within a clinical trial
-
The targeted therapy is non-standard because it was given in another therapy line, without a designated prior therapy or in combination with a different chemotherapy backbone, as specified by summary of product characteristics and/or treatment guidelines.
Data collection
Study sites are asked to retrospectively document all patients who meet the eligibility criteria (including deceased patients) until 2023.
Clinical routine data documented by study sites is structured and transferred by the site staff into a secure web-based eCRF system (iostudy office). The iostudy office edc system is a fully validated secure software that conforms to 21 CRF Part 11 requirements. As source of documentation, routine medical records are used. Sites will document data regarding patient and disease characteristics (e.g. sex, entity, staging, histology) as well as treatment details of NSTT and prior and subsequent therapies applied. The investigator’s treatment decision process is evaluated using a paper-based questionnaire comprising questions on factors influencing treatment decisions as well as information on respective potentially actionable alteration including ESCAT levels, which are transferred into the eCRF. eCRF documentation will be checked for plausibility and completeness both centrally and on-site by data managers, medical managers, and clinical monitors, respectively.
Study sites send pseudonymized diagnostic reports (e.g. pathology reports, reports on sequencing results such as NGS) and molecular tumor board (MTB) reports, if applicable, including treatment recommendations to iOMEDICO. Dedicated and trained iOMEDICO staff extracts information from the reports and enters data on all reported immunohistochemistry or mutational profiling as well as molecular alterations including complex sequencing results (including test panels and further information) into a pre-specified electronic database. These data are documented as described in the report but in analogy with the HGVS (Human genome variation society) code for proteomic und genomic sequence variants [23]. Independent checks for consistency of data entries by a second reviewer are performed. This is to ensure central quality control for these comprehensive and complex data. This structured data capture enables standardized and structured analyses.
Additionally, we have established a decentral tissue sample biobank where information on tumor samples including contact details of local pathology departments is captured. This enables us to request samples for upcoming translational research projects.
Descriptive analyses
Regarding patient and disease characteristics as well as therapies applied, the following data will be analyzed:
-
Patient and disease characteristics including e.g., sex, body mass index (BMI), Eastern Cooperative Oncology Group (ECOG) performance status, type of health insurance, comorbidities and tumors types, time from primary diagnosis to start of first NSTT, age at start of first NSTT
-
Details on NSTT including substance classes, treatment duration as well as prior and subsequent antineoplastic therapy lines
Characteristics regarding diagnostics and identified molecular alterations include the following:
-
Frequencies: type of molecular diagnostic testing performed, proteins and genes tested, multigene panels or NGS in molecular diagnostics, type (by panel size) of NGS library sequenced, altered proteins and genes, if tested, treatment recommendations given in molecular diagnostic reports, implemented treatment recommendations given in molecular diagnostic reports, use of a MTB, implementation of the treatment recommendation given by MTB
-
Clinical decision making: Frequency of answers rated with a Likert scale on patient’s non-suitability for standard therapy options / choice of performed molecular diagnostics / choice of selected molecular target and targeted non-standard therapy / reasons for the selection of targeted non-standard therapy / primary goal of the non-standard targeted therapy / expected advantages of the non-standard targeted therapy and frequency of ESMO Scale of Clinical Actionability for molecular Targets (ESCAT) evidence levels
-
Turnaround times:
-
◦ Duration from sample receipt to molecular testing result
-
◦ Duration from molecular testing result to start of NSTT
-
Effectiveness outcomes
Effectiveness of non-standard targeted therapy will be analyzed as follows:
-
Overall survival (OS)
-
Progression-free survival (PFS)
-
PFS ratio (i.e., PFS of non-standard targeted therapy divided by PFS of preceding therapy)
-
Time to treatment failure (TTF)
-
TTF ratio (i.e., TTF of non-standard targeted therapy divided by TTF of preceding therapy)
-
Tumor response: Best overall response (BOR); overall response rate (ORR); disease control rate (DCR); Time to Response (TTR); Duration of Response (DOR)
Statistical analysis
No formal sample size calculation was performed. The sample size of 500 patients has been chosen to enable subgroup analyses for e.g., specific tumor entities with a predefined minimum number of patients. For example, given the distribution of patients for specific tumor entities from the MOSCATO trial [8], we expect 50 patients or more in each of the following cancer entities: head and neck cancer, gastrointestinal cancer, lung cancer, breast cancer und genitourinary cancer. Furthermore, given 100 recruiting sites with 1-2 eligible patients per year and 4 documentation periods, it can reasonably be expected to reach the sample size of 500 patients in time.
All patients fulfilling the eligibility criteria and with documented start date of NSTT will be included in the full analysis set (FAS). All variables will be analyzed in a descriptive manner. Categorical variables will be presented as absolute and relative frequencies and continuous variables as number of observations, median and range. Time-to-event variables (PFS and OS) will be analyzed using the Kaplan-Meier method. PFS is defined as the time from start of therapy to date of documented tumor relapse/progression or death due to any cause, whichever comes first. OS is defined as the time from start of therapy to date of death due to any cause. Patients with no event will be censored at the date of last contact or start of subsequent systemic therapy. The PFS ratio will be calculated to compare PFS of NSTT with time to progression (TTP) of preceding antineoplastic therapy for each patient. BOR is defined as best documented response achieved under NSTT. DCR is defined as proportion of patients who achieved complete response (CR), partial response (PR) or stable disease (SD) as best response; ORR is defined as proportion of patients who achieved either CR or PR as best response. TTR is defined as time from start of treatment to first objective response observed for patients who achieved a CR or PR. DOR is defined as time from documentation of tumor response to disease progression or death from any cause. TTF is defined as time from start of therapy to discontinuation of treatment for any reason, including progression, toxicity, and death.
Patient subgroups based on different substance classes will be analyzed separately. Provided that numbers are sufficient, further subgroups will be analyzed, e.g. by tumor entity, biomarker, intervention. All subgroup analyses will be considered exploratory.
Interim analyses are planned to be conducted after each data collection phase (2020, 2021, 2022) and a final analysis will be conducted at the end of study (2023).
Statistical analyses will be performed using SAS software, version 9.4 or higher and R version 4.0.3 or higher.
Discussion
Physicians strive to identify the most appropriate and effective cancer treatment for each individual patient. However, reliable evidence for precision oncology approaches to guide physicians in their therapy decisions is still scarce. Thus, there is an urgent need to provide more evidence to determine relevant drug/alteration associations [24].
Only about 5% of cancer patients participate in clinical trials [25], often due to strict inclusion criteria [26]. With the increasing number of molecularly defined patient groups, the overall prevalence of patients with such alterations becomes even smaller, which makes recruitment into trials even more challenging. However, the element of randomization is still a very important technique in clinical research to investigate patient benefits and harms. The SHIVA trial as well as the ongoing international CUPISCO trial (NCT03498521) [27] are strong examples for the feasibility of randomization in precision oncology trials, supporting the conduction of randomized controlled trials in this research field. The goal of these two examples is not to test a specific drug against a standard of care, but to compare the whole approach of testing and recommended treatment against standard of care. Nevertheless, it is also of great importance to understand current practice of precision oncology approaches outside clinical trials. One of the major challenges in the field of precision oncology is the standardization of testing methods, their interpretation and actual implication for clinical decision making. These are research fields that can preferably be investigated in cohort studies with very broad inclusion criteria. In addition, data on effectiveness can easily be explored for hypothesis generation and non-randomized comparative analyses. Thus, establishing a registry study such as INFINITY will provide complementary evidence to the complex field of precision oncology leveraging data that combine patient’s clinical characteristics, the tumor genotype together with the treatment journey and clinical outcomes in real-world.
The non-interventional design of the INFINITY study represents both a limitation, as it precludes causal conclusions on differences between subgroups, and a strength because it allows presentation of real-world data on patients not selected by restrictive inclusion criteria and on test rates in routine care. The retrospective documentation is a potential risk of bias for reporting patients with a better response to NSTT by participating sites. However, this was mitigated by defining certain time periods for documentation. A strength of this study is the enrollment from both hospitals and office-based oncologists in private practices all over Germany, recruiting a large, representative study cohort.
In summary, the INFINITY registry aims to provide real-world data, including the clinical outcomes, on molecular alteration-matched targeted therapies used outside of their approved indication in patients with advanced solid tumors or hematologic malignancies. Thus, the results will describe how precision oncology is currently applied and implemented in routine clinical practice in Germany. Furthermore, the results will complement available evidence on effectiveness of precision oncology in routine clinical practice in general, and of specific drug class/alteration matches used outside their approved indications.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BOR:
-
Best overall response
- BRAF:
-
v-raf murine sarcoma viral oncogene homolog B1
- BRCA1/2:
-
Breast cancer 1/2
- DCR:
-
Disease control rate
- dMMR:
-
Mismatch repair deficient
- DOR:
-
Duration of response
- ECOG:
-
Eastern Cooperative Oncology Group
- eCRF:
-
Electronic case report form
- ERBB2:
-
Erb-b2 receptor tyrosine kinase 2
- FAS:
-
Full analysis set
- FDA:
-
Food and Drug Administration
- FISH:
-
Fluorescent in situ hybridization
- FLT-3:
-
FMS-like tyrosine kinase-3
- HER2:
-
Human epidermal growth factor receptor-2
- HGVS:
-
Human genome variation society
- IHC:
-
Immunohistochemistry
- MSI-H:
-
Microsatellite instability-high
- MTB:
-
Molecular tumor board
- NGS:
-
Next generation sequencing
- NSTT:
-
Non-standard targeted therapy
- NTRK:
-
Neurotrophic tyrosine receptor kinase
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- PD-L1:
-
Programmed death-ligand 1
- PFS:
-
Progression-free survival
- RET:
-
Rearranged during transfection
- TMB-H:
-
High tumor mutational burden
- TTF:
-
Time to treatment failure
- TTR:
-
Time to response
References
Kalia M. Personalized oncology: recent advances and future challenges. Metabolism. 2013;62:S11–4.
Tsimberidou AM, Fountzilas E, Nikanjam M, Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev. 2020;86:102019.
De Maria Marchiano R, et al. Translational research in the era of precision medicine: where we are and where we will go. J Pers Med. 2021;11:216.
Schlomm T, Rödiger T, Graalmann J. Präzisionsonkologie. Urol. 2021;60:3–7.
Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med. 2020;12:8.
Schwaederle M, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: a meta-analysis. JAMA Oncol. 2016;2:1452–9.
Haslem DS, et al. A Retrospective analysis of precision medicine outcomes in patients with advanced cancer reveals improved progression-free survival without increased health care costs. J Oncol Pract. 2017;13:e108–19.
Massard C, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7:586–95.
Tsimberidou A-M, et al. Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT): an MD Anderson Precision Medicine Study. JCO Precis Oncol. 2017;2017:PO.17.00002.
Haslem DS, et al. Precision oncology in advanced cancer patients improves overall survival with lower weekly healthcare costs. Oncotarget. 2018;9:12316–22.
Zhang Q, Fu Q, Bai X, Liang T. Molecular profiling-based precision medicine in cancer: a review of current evidence and challenges. Front Oncol. 2020;10:532403.
Crisci S, et al. Overview of current targeted anti-cancer drugs for therapy in onco-hematology. Med Kaunas Lith. 2019;55:E414.
Zhong L, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct Target Ther. 2021;6:201.
Stein MK, Oluoha O, Patel K, VanderWalde A. Precision medicine in oncology: a review of multi-tumor actionable molecular targets with an emphasis on non-small cell lung cancer. J Pers Med. 2021;11:518.
Food and Drug Administration (FDA). News release. March 16, 2023. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-dabrafenib-trametinib-pediatric-patients-low-grade-glioma-braf-v600e-mutation. Accessed 19 May 2023.
Food and Drug Administration (FDA). News release. September 21, 2022. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selpercatinib-locally-advanced-or-metastatic-ret-fusion-positive-non-small-cell-lung. Accessed 19 May 2023.
Le Tourneau C, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16:1324–34.
Hainsworth JD, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36:536–42.
Mangat PK, Halabi S, Bruinooge SS, Garrett-Mayer E, Alva A, et al. Rationale and design of the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. JCO Precis Oncol. 2018;2018:10.1200/PO.18.00122. https://doi.org/10.1200/PO.18.00122.
Gupta R, et al. Pertuzumab plus trastuzumab in patients with colorectal cancer with ERBB2 amplification or ERBB2/3 mutations: results from the TAPUR study. JCO Precis Oncol. 2022;6:306. https://doi.org/10.1200/PO.22.00306.
Al Baghdadi T, et al. Sunitinib in patients with metastatic colorectal cancer (mCRC) with FLT-3 amplification: results from the targeted agent and profiling utilization registry (TAPUR) study. Target Oncol. 2020;15:743–50.
Alva AS, et al. Pembrolizumab in patients with metastatic breast cancer with high tumor mutational burden: results from the targeted agent and profiling utilization registry (TAPUR) study. J Clin Oncol Off J Am Soc Clin Oncol. 2021;39:2443–51.
den Dunnen JT. Describing sequence variants using HGVS nomenclature. Methods Mol Biol Clifton NJ. 2017;1492:243–51.
Agarwala V, et al. Real-world evidence in support of precision medicine: clinico-genomic cancer data as a case study. Health Aff (Millwood). 2018;37:765–72.
Unger JM, Cook E, Tai E, Bleyer A. The role of clinical trial participation in cancer research: barriers, evidence, and strategies. Am Soc Clin Oncol Educ Book Am Soc Clin Oncol Annu Meet. 2016;35:185–98.
Harvey RD, et al. Impact of broadening trial eligibility criteria for patients with advanced non-small cell lung cancer: real-world analysis of select ASCO-friends recommendations. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27:2430–4.
Pauli C, et al. A Challenging task: identifying patients with Cancer of Unknown Primary (CUP) according to ESMO guidelines: the CUPISCO trial experience. Oncologist. 2021;26:e769–79.
Acknowledgements
The authors thank all patients, physicians and study teams participating in this study.
Funding
This work is supported by Bristol-Myers Squibb GmbH & Co KGaA (grant number N/A) and Roche Pharma AG (grant number N/A). Bristol-Myers Squibb GmbH & Co KGaA and Roche Pharma AG are not involved in study design, data collection and analysis, interpretation of results, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
FB, CV, NM, KP, SB, and SZ designed the study and developed the study protocol. SG wrote the first draft of the manuscript. CV, FB, KP, NM, BK and SZ edited the manuscript for intellectual content and contributed significantly to the manuscript. UM, JS, LS, SFG, MZ, TD, AS, and MS critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was registered at ClinicalTrials.gov, identifier NCT04389541, and received a positive consultation result by the Ethics Committee of the Landesärztekammer Baden-Württemberg, as well as local ethics committees of the participating physicians, if required, before start of patient enrollment. The study is conducted in accordance with the principles of the Declaration of Helsinki and all patients alive at the time of study registration provide written informed consent prior to inclusion into the study.
Consent for publication
Not applicable.
Competing interests
UM reports grants from the Dieter Schwarz Foundation; consulting fees by BMS, Sanofi, Pierre Fabre, and Roche; honoraria for lectures / presentations / manuscript writing by BMS; support for attending meetings and / or travel by Lilly, Amgen, Roche, BMS, and Pierre Fabre; participation on an Advisory Board for iOMEDICO; Chairmanship of Cancer Society Baden-Württemberg. JS reports support for the present manuscript by iOMEDICO; consulting fees by MSD; honoraria for lectures / presentations / manuscript writing by MSD; support for attending meetings and / or travel grants by Octapharm, GSK, and Ipsen; participation on a Data Safety Monitoring Board / Advisory Board for MSD, and Organon; honoraria from iOMEDICO, Celgene, Roche, Bristol-Myers Squibb, Clovis Oncology GmbH, GSK, Boehringer Ingelheim, Amgen, Novartis, MSD, AOP, Searchlight, Pharma Partner, Medixline GmbH, Eisai, HE Research GmbH, Octapharma, AbbVie, NIO, I+E Research, and Ipsen; membership of scientific organizations: Deutsche Krebsgesellschaft, Deutsche Gesellschaft für Hämatalogie und Onkologie, European Society of Medical Oncology, BNHO – Berufsverband der Niedergelassenen Hämatologen und Onkologen, WINHO – Wissenschaftliches Institut der Niedergelassenen Hämatologen und Onkologen, AKS – Arbeitskreis klinische Studien in onkologischen und hämatologischen Praxen, ASCO – American Society of Clinical Oncology. MZ reports honoraria for lectures / presentations / manuscript writing by Novartis, Roche, and Vifor; participation on a Data Safety Monitoring Board or Advisory Board by AbbVie, BMS, Celgene, Janssen, Novartis, Roche, and Vifor. TD reports participation on a Data Safety Monitoring Board / Advisory Board for Novartis as well as Steering Board participation for iOMEDICO. AS reports research grants from Celgene and Roche; honoraria from Amgen, AstraZeneca, Aurikamed, Bayer, Celgene, Clinsol, Connectmedica, Gilead, GSK, I-MED, Lilly, MCI Deutschland, Metaplan, MSD, Nanostring, Novartis, Onkowissen.de, Promedicis, Pfizer, Pierre Fabre, Roche, Seagen, Streamedup, Teva, Tesaro and Thieme; travel support from Celgene, Pfizer, and Roche. MS reports compensation for consultant activities by Amgen, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, Merck Serono, Novartis, Roche, Sanofi, and Takeda; honoraria for CME presentations by Amgen, Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, and Novartis; research funding to institution by AstraZeneca and Bristol-Myers Squibb. NM reports grants for trials and registries by Roche; Advisory Board consulting fees by Roche; honoraria for presentations by Roche, MSD, Eisai, and Lilly; travel grants by Roche. BK reports consulting fees by Astellas and Roche; Biontech stocks. FB, LS, SB, SFG, SG, SZ, KP, and CV declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Martens, U.M., Schröder, J., Bengsch, F. et al. The INFINITY study protocol: a retrospective cohort study on decision making and clinical impact of biomarker-driven precision oncology in routine clinical practice. BMC Cancer 23, 543 (2023). https://doi.org/10.1186/s12885-023-11046-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-11046-3