
Abstract
Background
Advanced gastro-oesophageal cancer (AGOC) carries a poor prognosis. No standard of care treatment options are available after first and second-line therapies. Regorafenib is an oral multi-targeted tyrosine kinase inhibitor targeting angiogenic, stromal, and oncogenic receptor tyrosine kinases. Regorafenib 160 mg daily prolonged progression free survival compared to placebo (INTEGRATE, phase 2). Regorafenib 80 mg daily in combination with nivolumab 3 mg/kg showed promising objective response rates (REGONIVO).
Methods/design
INTEGRATE II (INTEGRATE IIa and IIb) platform comprises two international phase III randomised controlled trials (RCT) with 2:1 randomisation in favor of experimental intervention. INTEGRATE IIa (double-blind) compares regorafenib 160 mg daily on days 1 to 21 of each 28-day cycle to placebo. INTEGRATE IIb (open label) compares REGONIVO, regorafenib 90 mg days 1 to 21 in combination with intravenous nivolumab 240 mg days 1 and 15 each 28-day cycle with investigator’s choice of chemotherapy (control). Treatment continues until disease progression or intolerable adverse events as per protocol. Eligible participants include adults with AGOC who have failed two or more lines of treatment. Stratification is by location of tumour (INTEGRATE IIa only), geographic region, prior VEGF inhibitor and prior immunotherapy use (INTEGRATE IIb only). Primary endpoint is overall survival. Secondary endpoints are progression free survival, objective response rate, quality of life, and safety. Tertiary/correlative objectives include biomarker and pharmacokinetic evaluation.
Discussion
INTEGRATE II provides a platform to evaluate the clinical utility of regorafenib alone, as well as regorafenib in combination with nivolumab in treatment of participants with refractory AGOC.
Trial registration
INTEGRATE IIa prospectively registered 1 April 2016 Australia New Zealand Clinical Trial Registry: ACTRN12616000420448 (ClinicalTrials.gov NCT02773524). INTEGRATE IIb prospectively registered 10 May 2021 ClinicalTrials.gov: NCT04879368.
Similar content being viewed by others


Background
Gastro-oesophageal cancer is the seventh most common cancer globally with an estimated 500,000 new cases diagnosed annually [1]. It is the sixth leading cause of cancer mortality globally [1]. Despite current treatments, advanced gastro-oesophageal cancer (AGOC) carries a poor prognosis, with a median overall survival (OS) of less than 12 months [2].
Chemotherapy is a well-established first- and second-line treatment option for patients with AGOC. The combination of fluoropyrimidine and platinum-based chemotherapy is commonly used in the first line setting in patients with human epidermal growth factor receptor 2 (HER2) negative AGOC [3]. Trastuzumab is added to doublet chemotherapy in patients with HER2 positive AGOC [3]. First line chemotherapy in AGOC improves OS compared to best supportive care (HR 0.37, 95% CI 0.24–0.55), with longer OS seen with doublet chemotherapy compared to single agent chemotherapy (HR 0.82, 95% CI 0.74–0.90) [3]. In the second-line setting, commonly used regimens include chemotherapy (taxanes or irinotecan) [4,5,6] ramucirumab as monotherapy, and ramucirumab in combination with chemotherapy [7]. Second-line chemotherapy also improves OS in patients with AGOC compared to best supportive care (HR 0.73, 95% CI 0.58–0.96) [8]. In the third or later line setting, trifluridine/tipiracil improves median OS by 5.7 months compared to 3.6 months placebo in the TAGS trial (HR 0.69, 95% CI 0.56–0.85) [9]. In the phase III ATTRACTION-2 study, nivolumab improves median OS when compared to placebo in participants with AGOC who were refractory or intolerant to two or more lines of chemotherapy (HR 0.63, 95% CI 0.51–0.78) [10].
Regorafenib (BAY 73–4506) is a promising treatment option in AGOC. Regorafenib is an oral multikinase inhibitor (TKI) that targets angiogenic (VEGF, TIE-2), stromal (PDGF-β), and oncogenic (RAF, RET and KIT) receptor tyrosine kinases [11]. Regorafenib showed promise in the treatment of participants with chemotherapy refractory AGOC in the INTEGRATE trial [12]. INTEGRATE was an international phase II multi-centre Randomised Controlled Trial (RCT), sponsored by the Australasian Gastro-Intestinal Trials Group (AGITG), comprising of 152 participants with AGOC who had progressed on first- and/or second-line chemotherapy [12]. INTEGRATE demonstrated an improved progression free survival (PFS) with regorafenib at a dose of 160 mg daily on days 1 to 21 of a 28-day cycle compared to placebo (HR 0.40, 95% CI 0.28–0.59, p < 0.001) [12]. There was a trend towards an OS benefit with regorafenib (HR 0.74, 95% CI 0.51–1.08, p = 0.147), but the study was underpowered to reliably detect a plausible OS effect and 58% of placebo participants crossed over to receive regorafenib following disease progression [12]. Pre-specified analyses demonstrated a higher efficacy of regorafenib amongst participants in Korea compared to participants in Australia, New Zealand, and Canada (HR 0.12 vs HR 0.61, p < 0.001) suggesting a difference in progression-free survival by ethnicity/geographical region [8]. There were no unexpected toxicities with regorafenib [12].
The results of INTEGRATE formed the basis for launching the original INTEGRATE II phase III RCT with the aim of investigating whether regorafenib at a dose of 160 mg daily given on days 1 to 21 of a 28-day cycle is better than placebo in prolonging OS in participants with refractory AGOC who have failed two or more lines of chemotherapy. Both arms received best supportive care and crossover of placebo participants to receive regorafenib at disease progression was not allowed.
After INTEGRATE II opened for recruitment, standard practice in the management of AGOC shifted with increasing evidence through phase II [13, 14] and phase III RCTs supporting the use of immune checkpoint inhibitors in first and later line treatment [10, 15]. For example, the combination of nivolumab plus chemotherapy in CheckMate 649 resulted in a significant improvement in OS (HR 0.71, 98% CI 0.59–0.86, p < 0.0001) and PFS (HR 0.68, 98% CI 0.56–0.81, p < 0.0001) in participants with AGOC and a PD-L1 combined positive score (CPS) score of 5 or more in the first line setting [15]. Although the combination of upfront chemotherapy and immune checkpoint inhibitors is suggested to be superior to chemotherapy alone in the first line treatment of AGOC [15], most patients will eventually experience disease progression despite aggressive upfront therapies. This highlighted the need for the evaluation of newer combination therapies to overcome primary and acquired resistance to current therapies.
The INTEGRATE II research plan was consequently expanded to become a platform protocol that included a new second trial comparing regorafenib in combination with nivolumab (experimental treatment) against chemotherapy (control treatment). This new study was called INTEGRATE IIb and the original study was renamed INTEGRATE IIa.
INTEGRATE IIb is a phase III RCT that investigates whether regorafenib 90 mg daily on day 1 to 21 of a 28-day cycle in combination with nivolumab 240 mg every 2 weeks is superior to chemotherapy alone in prolonging OS in the overall and Asian sub-population of participants with AGOC who have progressed following two or more lines of treatment. The rationale behind INTEGRATE IIb is the REGONIVO phase Ib trial, a dose-finding/dose expansion Japanese study which assessed the combination of regorafenib and nivolumab in 50 participants with previously treated gastric and colorectal cancers [16]. As part of the dose expansion study, regorafenib was given at a dose of 80 mg to 160 mg once daily on days 1 to 21 of a 28-day cycle together with nivolumab 3 mg/kg every 2 weeks until disease progression or dose limiting toxicities [16]. The authors of REGONIVO concluded that the optimal dose of regorafenib when given in combination with nivolumab 3 mg/kg every 2 weeks was 80 mg daily on days 1 to 21 of a 28-day cycle [16]. The overall response rate (ORR) in the REGONIVO study was 44% in microsatellite stable (MSS) gastric cancer and 33% in MSS colorectal cancer [16]. These impressive ORR results provided a rationale for further investigation of this combination therapy in larger cohorts [16].
INTEGRATE IIa and INTEGRATE IIb are two relevant trials that will further clarify the role of regorafenib in the treatment of AGOC. INTEGRATE IIa will answer the important question of whether regorafenib monotherapy is better than placebo in participants with chemotherapy refractory AGOC. INTEGRATE IIa will also provide information on the role of regorafenib monotherapy in participants who cannot have further chemotherapy or immune checkpoint inhibitors. INTEGRATE IIb seeks to evaluate the efficacy of regorafenib in combination with immune checkpoint inhibitors in comparison to standard of care chemotherapy in the third or later line setting.
Methods/ Design
Aim
The aim of INTEGRATE IIa is to determine whether regorafenib160mg daily on day 1 to 21 of a 28-day cycle improves OS compared to placebo. The aim of INTEGRATE IIb is to determine whether regorafenib 90 mg daily on day 1 to 21 of a 28-day cycle in combination with nivolumab 240 mg every 2 weeks improves OS compared to investigator’s choice of chemotherapy in participants with AGOC who have progressed or were intolerant to two or more lines of chemotherapies including platinum and fluoropyrimidine based regimens. After 2 months, nivolumab can be given monthly at a dose of 480 mg.
Design
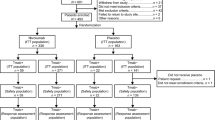
INTEGRATE IIa is a prospective, multi-centre, comparative, double-blinded phase III RCT where participants with histological or cytological confirmation of AGOC or undifferentiated carcinoma of primary gastro-oesophageal origin are randomised 2:1 to receive either regorafenib monotherapy (Arm A) or placebo (Arm B) (Fig. 1). Randomisation is stratified by location of tumour (GOJ (gastro-oesophageal junction) vs. gastric), geographic region (Asia vs. rest of world) and prior VEGF inhibitors (yes vs no).

Schema of INTEGRATE IIa
INTEGRATE IIb is a prospective, multi-centre, comparative, open label phase III RCT where participants with histological or cytological confirmation of AGOC or undifferentiated carcinoma of primary gastro-oesophageal origin are randomised 2:1 to receive regorafenib and nivolumab (experimental arm) or investigator’s choice of chemotherapy (control arm) (Fig. 2). Randomisation is stratified by geographic region (Asia vs. rest of world), prior VEGF inhibitors (yes vs no) and prior immunotherapy (yes vs no).

Schema of INTEGRATE IIb
For INTEGRATE IIa and INTEGRATE IIb, all treatments are to be administered until disease progression, unacceptable toxicity, withdrawal of consent, more than 2 dose reductions due to treatment related adverse events or day 1 treatment delays of longer than 28 days. Randomisation will be performed centrally. Participant quality of life (QOL) questionnaires are to be completed on cycle 1 day 1 of treatment and every 4 weeks thereafter during treatment and at the time of disease progression. Participants who end treatment before disease progression will have QOL data collected every 8 weeks until disease progression. A list of participating centres is provided in Table 1 for INTEGRATE IIa and Table 2 for INTEGRATE IIb.
Study endpoints
The primary end point for INTEGRATE IIa and INTEGRATE IIb is overall survival in each arm, defined as the interval from date of randomisation to date of death from any cause or the date the patient was last known to be alive. Secondary objectives for INTEGRATE IIa and INTEGRATE IIb are PFS, ORR (according to RECIST v1.1), QOL and safety profile (rates of adverse events per CTCAE v4.03). The tertiary and correlative endpoints may include: investigation of VEGF-related biomarkers and biomarkers relating to angiogenesis and/or tumourigenesis in blood and tumour as prognostic and/or predictive markers for study endpoints relating to survival, response and safety; regorafenib levels to evaluate pharmacokinetics in participants from different geographical regions; evaluation of the prevalence and distribution of the four proposed molecular phenotypes of gastric cancer proposed by the Cancer Genome Atlas Research Network [17], and their association with angiogenic biomarkers and regorafenib activity. Additional correlative endpoints for INTEGRATE IIb may include associations between autoimmunity and clinical outcomes, immune profiling of tumour, tumour mutational burden and cellular and molecular signatures associated with immune related adverse events.
Study population
INTEGRATE IIa and INTEGRATE IIb have similar eligibility criteria:
Participants aged ≥ 18 years with ECOG performance status < 2, who have histological or cytological confirmation of metastatic or locally advanced gastro-oesophageal carcinoma (gastric or GOJ cancer). Participants must have failed or be intolerant to a minimum of 2 lines of prior anti-cancer therapy and must have had at least one platinum agent and one fluoropyrimidine analogue.
Other main eligibility criteria include:
-
1. Evaluable disease according to RECIST 1.1
-
2. Adenocarcinoma or undifferentiated carcinoma histology
-
3. Neoadjuvant or adjuvant chemotherapy or chemoradiotherapy will be considered as first line treatment if the patient has relapsed or progressed within 6 months of completing treatment
-
4. Ramucirumab monotherapy or immunotherapy with a checkpoint inhibitor will be considered a line of treatment
-
5. HER2 positive participants must have previously received trastuzumab
-
6. Adequate organ function defined as below:
-
i. Bone marrow: ANC (absolute neutrophil count) ≥ 1500/μl, platelets ≥ 100,000/μl, haemoglobin ≥ 9 g/dl. INR (international normalised ratio) and APTT (activated partial thromboplastin time) ≤ 1.5 × ULN (upper limit of normal). Note: participants previously on long-term anticoagulation with warfarin or low molecular weight heparin are eligible.
-
ii. Adequate liver function: Total bilirubin ≤ 1.5 × ULN; AST (aspartate transaminase), ALT (alanine transaminase) and/or ALP (alkaline phosphatase) ≤ 5 × ULN.
-
iii. Adequate renal function, creatinine clearance, as measured by the Cockcroft and Gault formula of > 50mls/minute.
-
iv. Adequate cardiac function: left ventricular ejection fraction ≥ 50% (INTEGRATE IIa only).
-
-
7. Study treatment to start within 7 days of randomisation
Main exclusion criteria include:
-
1. Poorly controlled hypertension (systolic blood pressure > 140 mmHg or diastolic pressure > 90 mmHg despite optimal medical management)
-
2. Prior exposure to anti-VEGF targeted therapy using small molecule VEGF TKIs. Prior anti-VEGF targeted monoclonal antibody therapies such as bevacizumab and ramucirumab are permitted
-
3. Any prior use of more than one immune checkpoint inhibitor (INTEGRATE IIb only)
-
4. Palliative radiotherapy unless more than 14 days between completion of radiation and date of registration and adverse events resulting from radiation have resolved to less than grade 2 according to CTCAE V4.03
-
5. Interstitial lung disease with ongoing signs and symptoms
-
6. Uncontrolled metastatic disease to central nervous system
Study interventions
In INTEGRATE IIa, participants enrolled in Arm A will receive regorafenib 160 mg (4 × 40 mg tablets) orally once daily on days 1 to 21 of a 28-day cycle until disease progression, unacceptable toxicities, day 1 treatment delay of more than 28 days or withdrawal of consent. Participants enrolled in Arm B will receive matching placebo. Missed or vomited tablets cannot be compensated for by treatment later.
In INTEGRATE IIb, participants enrolled in the experimental arm of the study will receive regorafenib and nivolumab. Regorafenib will be self-administered orally once daily at a dose of 90 mg (3 × 30 mg tablets) on days 1 to 21 of a 28-day cycle until disease progression, unacceptable toxicities, day 1 treatment delay of more than 28 days or withdrawal of consent. Only one dose level reduction of regorafenib is permitted (90 mg to 60 mg) for management of regorafenib related adverse events. If regorafenib is not tolerated at a dose of 60 mg, regorafenib will be discontinued. Nivolumab will be administered intravenously at a dose of 240 mg on day 1 of a 14-day cycle until disease progression or prohibitive adverse events as per protocol or up to 24 infusions or 2 years maximum treatment duration. After 2 months, nivolumab can be administered at a dose of 480 mg on day 1 of a 28-day cycle. If a delay in nivolumab administration is required when a treatment cycle has commenced, nivolumab cannot be delayed more than 12 days for the 14-day schedule, and not more than 20 days for the 28-day schedule. If nivolumab needs to be delayed more than 12 weeks from last treatment date due to toxicity, nivolumab will be discontinued permanently. The continuation of regorafenib monotherapy is permitted despite permanent discontinuation of nivolumab. Participants enrolled in the control arm will receive investigator’s choice of chemotherapy with either docetaxel 60-75 mg/m2 intravenously every 21 days, or paclitaxel 135-250 mg/m2 intravenously on day 1 every 21 days, or paclitaxel 80 mg/m2 intravenously on days 1, 8, 15, 21 every 28 days, or irinotecan 250-300 mg/m2 intravenously on day 1 every 21 days, or irinotecan 150-180 mg/m2 intravenously on days 1 and 15 every 28 days, or irinotecan 125 mg/m2 intravenously on days 1 and 8 every 28 days, or trifluridine and tipiracil 35 mg/m2 up to 80 mg dose orally twice daily on days 1 to 5 and 8 to 12 of a 28 day cycle.
All participants in INTEGRATE IIa and INTEGRATE IIb will receive best supportive care which includes any intervention to preserve comfort and dignity such as stent insertion for gastric outlet obstruction, radiation to a bleeding primary tumour and use of any concomitant medications considered necessary for participant’s wellbeing including antiemetics, antidiarrhoeals, anti-inflammatory agents and analgesics.
Study assessments
Disease assessment by CT scan will be performed at baseline and every 8 weeks, regardless of delays in drug administration until disease progression. QOL questionnaire (European Organization for Research and Treatment of Cancer (EORTC) QLQ-C30 and EORTC QLQ-STO22) will be completed at baseline and every 4 weeks during treatment and at the time of disease progression. For participants who end treatment before disease progression, QOL questionnaire will be collected every 8 weeks during follow up until disease progression. Tumour marker CA19-9 is assessed at the same time as CT imaging until disease progression.
Translational research
Blood for biomarker research is collected from all participants at four timepoints: Cycle 1 Day 1, Cycle 2 Day 1, Cycle 4 Day 1, and end of treatment. Blood for pharmacokinetics (PK) is collected at three timepoints: Cycle 1 Day 15, Cycle 2 Day 1, and Cycle 2 Day 15 (optional) drawn before regorafenib is taken and 1 to 4 h afterwards. All blood samples collected are processed, frozen and biobanked. Archival tissue from the primary tumour or metastases are retrieved for translational research.
Statistical considerations
The primary analysis for INTEGRATE IIa and IIb is a comparison of OS between the experimental and control arms using a log-rank test accounting for stratification factors. A HR for OS will be estimated from a stratified Cox proportional hazards model accounting for stratification factors. PFS will be analysed in a comparable fashion to OS. ORR will be compared using Cochran-Mantel–Haenszel test accounting for stratification factors or Fisher’s exact test if expected cell counts are low. Quality of life will be analysed using the approaches applied to the INTEGRATE trial [18]. A series of subgroup analyses will be performed on OS for the stratification factors used at randomisation.
When the INTEGRATE platform protocol was proposed, the original sample size target for INTEGRATE II (regorafenib vs placebo) of N = 350 was changed to N = 250 for the INTEGRATE IIa trial. A study with 250 participants randomised in a 2:1 ratio (regorafenib: placebo) to yield an expected 221 OS events and have 80% power to detect a HR for OS of 0.67 with a 2-sided α of 5%. The OS events from INTEGRATE IIa will also contribute to a pooled analysis with the previous INTEGRATE trial which had 123 OS events. The conditional power of this combined pooled analysis will have at least 90% power to detect a HR for OS of 0.67. To determine the appropriateness of a pooled analysis, heterogeneity of treatment effect on OS across INTEGRATE and INTEGRATE IIa will be tested. Assuming there is no statistically significant heterogeneity, a sequential closed testing procedure (with a 2-sided α of 5%) will be applied to the following null hypotheses of no treatment effect in the:
-
• Pooled cohort [INTEGRATE + INTEGRATE IIa] (\({\mathrm{H}0}_{1}\))
-
• INTEGRATE IIa cohort (\({\mathrm{H}0}_{2}\))
-
• Asian region pooled cohort [INTEGRATE + INTEGRATE IIa] (\({\mathrm{H}0}_{3}\))
-
• Asian region INTEGRATE IIa cohort (\({\mathrm{H}0}_{4}\))
For INTEGRATE IIb, a sample of 450 participants will be randomised 2:1 (regorafenib and nivolumab: chemotherapy) and followed until 380 deaths occur to provide at least 90% power to detect a HR for OS of 0.70 with a 2-sided α of 0.05. The design also accommodates early stopping for benefit/harm at an interim analysis performed at 2/3 of the required events using the error spending approach of Lan-DeMets. A safety analysis will be undertaken when a minimum of 8 participants are enrolled from Japan, 16 from Asia and the remainder from USA, Europe and Australasia. The final analysis on OS will involve testing the following two null hypotheses: (1) no treatment effect on OS in the whole trial cohort (\({H}_{0}^{All}\)); and, (2) no treatment effect on OS in the Asian region cohort (\({H}_{0}^{Asian}\)). A sequenced closed testing procedure will be used to constrain the overall type I error of these two tests to 5%.
Safety
All adverse events for INTEGRATE IIa and INTEGRATE IIb will be recorded from the first dose of study treatment until 30 days (90 days for nivolumab) following last treatment dose. The investigator is responsible for ensuring all adverse events observed by the investigator or reported by the trial participants are documented in electronic case report forms (eCRFs). Serious adverse events (SAEs), including suspected unexpected serious adverse reactions (SUSAR) occurring during the study must be reported to the sponsor within 24 h of investigational site staff becoming aware of the event according to local procedures. The sponsor is responsible for the medical review of all SAEs and for their notification to the appropriate ethics committees and local authorities.
Discussion
INTEGRATE IIa and INTEGRATE IIb address the clinically important question of exploring later line therapies that can improve overall survival in participants with refractory AGOC. INTEGRATE IIa and INTEGRATE IIb have similar inclusion/exclusion criteria and primary/secondary endpoints. The use of one previous line of immune checkpoint inhibitor is allowed in INTEGRATE IIb. The rationale for this is that some participants may have had immunotherapy in combination with chemotherapy as first line treatment for their AGOC as per the CheckMate 649 study [15]. Another rationale for this is that the combination of regorafenib and nivolumab in the REGONIVO study showed promising objective response rates [16].
There are a few major differences between INTEGRATE IIa and INTEGRATE IIb. INTEGRATE IIa compares regorafenib to placebo whilst INTEGRATE IIb compares regorafenib and nivolumab to chemotherapy. INTEGRATE IIa uses placebo as the control arm as there was minimal evidence at the time of protocol development to support third line chemotherapy as the standard of care. However, a systematic review and meta-analysis by Zheng et al. from 19 studies subsequently showed that third-line chemotherapy compared to placebo was associated with a better median OS (HR 0.68, 95% CI 0.57–0.82, p < 0.001) and progression-free survival (HR 0.56, 95% CI 0.44–0.71, p > 0.01) in participants with AGOC [19]. A randomised phase 3 study of trifluridine/tipiracil versus placebo also demonstrated an OS advantage in this pre-treated population (HR 0.69, 95% CI 0.56–0.85), p < 0.001) [9]. As such, it was decided that INTEGRATE IIb should use chemotherapy as the control arm.
Another difference between INTEGRATE IIa and INTEGRATE IIb is the starting dose of regorafenib. In INTEGRATE IIa, the starting dose of regorafenib is 160 mg (4 × 40 mg) whilst the starting dose of regorafenib is lower in INTEGRATE IIb 90 mg (3 × 30 mg). In the REGONIVO phase I study, the optimal dose of regorafenib when combined with nivolumab in terms of efficacy and safety is 80 mg [16]. However, since completion of the REGONIVO study, Bayer has developed a 30 mg tablet formulation dose for regorafenib that is expected to provide very similar exposure to 80 mg due to PK variability and will permit a dose reduction to 60 mg using 2 × 30 mg tablets dose level -1 = 33% reduction [20].
In summary, INTEGRATE II provides a platform to evaluate the clinical utility of regorafenib alone, as well as regorafenib in combination with nivolumab in the treatment of participants with AGOC who have failed two or more lines of treatment. The results of this study will hopefully improve the current standard of care in the treatment of AGOC.
Availability of data and materials
Not applicable.
Abbreviations
- AGOC:
-
Advanced gastro-oesophageal cancer
- HER2:
-
Human epidermal growth factor receptor 2
- TKI:
-
Tyrosine kinase inhibitor
- OS:
-
Overall survival
- PFS:
-
Progression free survival
- QOL:
-
Quality of life
- ORR:
-
Objective response rate
- CPS:
-
Combined positive score
- PK:
-
Pharmacokinetics
- ALP:
-
Alkaline phosphatase
- ANC:
-
Absolute neutrophil count
- APTT:
-
Activated partial thromboplastin time
- AST:
-
Aspartate transaminase (or aspartate aminotransferase)
- CTCAE:
-
Common terminology criteria for adverse events
- RECIST:
-
Response evaluation criteria in solid tumours
- ULN:
-
Upper limit of normal
- VEGF:
-
Vascular endothelial growth factor
- TIE-2:
-
Tyrosine kinase with immunoglobulin and EGF homology domains
- PDGF-β:
-
Platelet derived growth factor receptor—β
- RAF:
-
Rapidly accelerated fibrosarcoma (raf) kinase
- RET:
-
Ret proto-oncogene
- KIT:
-
Proto-oncogene c-KIT
- IDSMC:
-
Independent data and safety monitoring committee
- eCRFs:
-
Electronic case report forms
- HR:
-
Hazard ratio
- SAEs:
-
Serious adverse events
- SUSAR:
-
Suspected unexpected serious adverse events
- NHMRC:
-
National health and medical research council clinical trials centre
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clin. 2018;68:394–424.
Abbas MN, Bright T, Price T, Karapetis C, Thompson S, Connell C, Watson D, Barnes M, Bull J, Singhal N, Roy A. Patterns of care and outcomes for gastric and gastro-oesophageal junction cancer in an Australian population. ANZ J Surg. 2021;91(12):2675–82.
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Rüschoff J, Kang YK. ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–97.
Lee KW, Maeng CH, Kim TY, Zang DY, Kim YH, Hwang IG, Oh SC, Chung JS, Song HS, Kim JW, Jeong SJ, Cho JY. A Phase III Study to Compare the Efficacy and Safety of Paclitaxel Versus Irinotecan in Patients with Metastatic or Recurrent Gastric Cancer Who Failed in First-line Therapy (KCSG ST10-01). Oncologist. 2019;24(1):18-e24.
Hironaka S, Ueda S, Yasui H, Nishina T, Tsuda M, Tsumura T, Sugimoto N, Shimodaira H, Tokunaga S, Moriwaki T, Esaki T, Nagase M, Fujitani K, Yamaguchi K, Ura T, Hamamoto Y, Morita S, Okamoto I, Boku N, Hyodo I. Randomized, open-label, phase III study comparing irinotecan with paclitaxel in patients with advanced gastric cancer without severe peritoneal metastasis after failure of prior combination chemotherapy using fluoropyrimidine plus platinum: WJOG 4007 trial. J Clin Oncol. 2013;31(35):4438–44.
Ford HE, Marshall A, Bridgewater JA, Janowitz T, Coxon FY, Wadsley J, Mansoor W, Fyfe D, Madhusudan S, Middleton GW, Swinson D, Falk S, Chau I, Cunningham D, Kareclas P, Cook N, Blazeby JM, Dunn JA. COUGAR-02 Investigators. Docetaxel versus active symptom control for refractory oesophagogastric adenocarcinoma (COUGAR-02): an open-label, phase 3 randomised controlled trial. Lancet Oncol. 2014;15(1):78–86.
Young K, Smyth E, Chau I. Ramucirumab for advanced gastric cancer or gastro-oesophageal junction adenocarcinoma. Therap Adv Gastroenterol. 2015;8(6):373–83.
Iacovelli R, Pietrantonio F, Farcomeni A, Maggi C, Palazzo A, Ricchini F, et al. Chemotherapy or targeted therapy as second-line treatment of advanced gastric cancer. A systematic review and meta-analysis of published studies. PLoS One. 2014;9(9):e108940.
Shitara K, Doi T, Dvorkin M, Mansoor W, Arkenau HT, Prokharau A, Alsina M, Ghidini M, Faustino C, Gorbunova V, Zhavrid E, Nishikawa K, Hosokawa A, Yalçın Ş, Fujitani K, Beretta GD, Cutsem EV, Winkler RE, Makris L, Ilson DH, Tabernero J. Trifluridine/tipiracil versus placebo in patients with heavily pretreated metastatic gastric cancer (TAGS): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19(11):1437–48.
Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, Chung HC, Chen JS, Muro K, Kang WK, Yeh KH, Yoshikawa T, Oh SC, Bai LY, Tamura T, Lee KW, Hamamoto Y, Kim JG, Chin K, Oh DY, Minashi K, Cho JY, Tsuda M, Chen LT. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10111):2461–71.
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, Thierauch KH, Zopf D. Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–55.
Pavlakis N, Sjoquist KM, Martin AJ, et al. Regorafenib for the Treatment of Advanced Gastric Cancer (INTEGRATE): A Multinational Placebo-Controlled Phase II Trial. J Clin Oncol. 2016;34(23):2728–35.
Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, Sun W, Jalal SI, Shah MA, Metges JP, Garrido M, Golan T, Mandala M, Wainberg ZA, Catenacci DV, Ohtsu A, Shitara K, Geva R, Bleeker J, Ko AH, Ku G, Philip P, Enzinger PC, Bang YJ, Levitan D, Wang J, Rosales M, Dalal RP, Yoon HH. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018;4(5):e180013.
Janjigian YY, Bendell J, Calvo E, Kim JW, Ascierto PA, Sharma P, Ott PA, Peltola K, Jaeger D, Evans J, de Braud F, Chau I, Harbison CT, Dorange C, Tschaika M, Le DT. CheckMate-032 Study: Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Patients With Metastatic Esophagogastric Cancer. J Clin Oncol. 2018;36(28):2836–44.
Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, Wyrwicz L, Yamaguchi K, Skoczylas T, Campos Bragagnoli A, Liu T, Schenker M, Yanez P, Tehfe M, Kowalyszyn R, Karamouzis MV, Bruges R, Zander T, Pazo-Cid R, Hitre E, Feeney K, Cleary JM, Poulart V, Cullen D, Lei M, Xiao H, Kondo K, Li M, Ajani JA. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398(10294):27–40.
Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, Yoshii T, Kotani D, Tamura H, Mikamoto Y, Hirano N, Wakabayashi M, Nomura S, Sato A, Kuwata T, Togashi Y, Nishikawa H, Shitara K. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38(18):2053–61.
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50.
Martin AJ, Gibbs E, Sjoquist K, Pavlakis N, Simes J, Price T, Shannon J, Gill S, Jain V, Liu G, Kannourakis G, Kim YH, Kim JW, Goldstein D. INTEGRATE I investigators. Health-related quality of life associated with regorafenib treatment in refractory advanced gastric adenocarcinoma. Gastric Cancer. 2018;21(3):473–80.
Zheng Y, Zhu XQ, Ren XG. Third-line chemotherapy in advanced gastric cancer: a systematic review and meta-analysis. Medicine. 2017;96(24):e6884.
El-Khoueiry AB, Kim RD, Harris WP, Sung MW, Waldschmidt D, Iqbal S, et al. Phase Ib study of regorafenib (REG) plus pembrolizumab (PEMBRO) for first-line treatment of advanced hepatocellular carcinoma (HCC). American Society of Clinical Oncology; 2020.
Acknowledgements
This study is sponsored by the Australasian Gastro-Intestinal Trials Group (AGITG) and conducted in collaboration with NHMRC Clinical Trial Centre, University of Sydney, Australia. The study is supported by Bayer HealthCare Pharmaceuticals and Bristol Myers Squibb. We thank the participants, their families and the investigators who have made this study possible.
Role of funding source
Bayer HealthCare Pharmaceuticals and Bristol Myers Squibb had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results. Bayer HealthCare Pharmaceuticals and Bristol Myers Squibb have the right to review and give feedback prior to submitting a manuscript.
Funding
INTEGRATE II is supported by an untied educational grant from Bayer HealthCare Pharmaceuticals to the Australasian Gastro-Intestinal Trials Group to conduct the study independently. Bayer HealthCare Pharmaceuticals are also providing the study drug regorafenib. Bristol Myers Squibb are providing the study drug nivolumab.
Author information
Authors and Affiliations
Contributions
LLL was involved in drafting the manuscript. NP, KS, KMS, AM, SY, YK, YB, LC, MM, TBS, TA, CO, NT, HC, SR, KL, JZ, TP, JS and DG have made substantial contribution to conception and design and helped to draft the manuscript. AJ and WH provided central coordination, management and helped to draft the manuscript. SY designed translational sub studies and helped draft the manuscript. AM and VG were involved in the statistical consideration of the study and helped draft the manuscript. NP, KS, KMS, YK, YB, LC, MM, TBS, TA, CO, NT, HC, SR, KL, JZ, TP, JS, DG provided study data through recruitment of patients. LLL, NP, KMS, NT, JZ, DG, TP, SY and AJ are members of the Trial Management Committee. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
INTEGRATE II (Protocol v7.0, 27th November 2020) was approved by the Human Research Ethics Committee (Royal North Shore Hospital) of the Northern Sydney Local Health District (NSLHD) as well as individual institutional ethics committees for sites not under NSLHD central ethics approval. The study is performed in accordance with the NHMRC National Statement on Ethical Conduct in Research Involving Humans 2007 (updated 2018), the NHMRC Australian Code for Responsible Conduct of Research 2018 and the principles laid down by the World Medical Assembly in the Declaration of Helsinki 2008. All participants must provide written informed consent to the study procedures before enrolment in the study.
Consent for publication
Not applicable. No data reported.
Competing interests
A potential competing interest is the provision of study drug regorafenib by BAYER.
Kohei Shitara reports receiving personal fees for advisory roles from Lilly, Bristol Myers Squibb, Takeda, Pfizer, Ono Pharmaceutical, Merck Pharmaceutical, Taiho Pharmaceutical, Astellas, Novartis, AbbVie, GlaxoSmithKline, Daiichi Sankyo, Amgen, Boehringer Ingelheim, Guardant Health Japan, and Janssen; receiving honoraria (lecture fee) from Takeda, Bristol-Myers Squibb and Janssen; and receiving research funding from Astellas, Ono Pharmaceutical, Daiichi Sankyo, Taiho Pharmaceutical, Chugai, Merck Pharmaceutical, Medi Science, Eisai and Amgen, outside the submitted work.
Keun-Wook Lee reports receiving grants from Ono pharmaceutical, AstraZeneca, Merck Sharp and Dohme, Merck KGaA, Pfizer, BeiGene, Zymeworks, ALX Oncology, Astellas, Macrogenics, Five Prime Therapeutics, Seagen, Bolt therapeutics, Trishula therapeutics, Oncologie, Pharmacyclics, Daiichi Sankyo, Taiho Pharmaceutical, InventisBio, Leap therapeutics, MedPacto, LSK BioPharma, Green Cross Corp, ABLBIO, Y-BIOLOGICS, Genexine, Metafines (to his institution for conducting clinical trials), personal fees from Metafines, Bayer, Daiichi Sankyo, Merck Sharp and Dohme, Bristol Myers Squibb, Vifor Pharma (consultation) and Ono pharmaceutical, Boryung (honorarium) outside the submitted work.
Thierry Alcindor reports receiving consulting/advisory fees from Merck, Bayer, BMS, Astrazeneca, Astellas Scientific and Medical Affairs Inc and research funding from epizyme, emd serono, karyopharm therapeutics, springworks therapeutics, astellas pharma and deciphera.
Tanios Bekaii-Saab reports receiving research funding to institution from Agios, Arys, Arcus, Atreca, Boston Biomedical, Bayer, Eisai, Celgene, Lilly, Ipsen, Clovis, Seattle Genetics, Genentech, Novartis, Mirati, Merus, Abgenomics, Incyte, Pfizer, BMS; Consulting fees to institution from Ipsen, Arcus, Pfizer, Seattle Genetics, Bayer, Genentech, Incyte, Eisai, Merus, Merck KGA and Merck; personal consulting fees from Stemline, AbbVie, Boehringer Ingelheim, Janssen, Daiichi Sankyo, Natera, TreosBio, Celularity, Caladrius Biosciences, Exact Science, Sobi, Beigene, Kanaph, Astra Zeneca, Deciphera, Zai Labs, MJH Life Sciences, Aptitude Health, Illumina and Foundation Medicine.
Katrin Marie Sjoquist reports receiving honororia from AMGEN, BMS, Ipsen, Merck and SERVIER; research funding to institution from Bayer; consulting/advisory role for Competitive Drug Development International.
Yung-Jue Bang reports consulting on the advisory board for MSD, Merck Serano, Daiichi-Sankyo, Astellas, Alexo Oncol, Samyang Biopharm, Hanmi, Daewoong and Amgen; receiving research grants from Genentech/Roche, MSD, Merck Serano, Daiichi Sankyo, Astellas and Amgen.
The other authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Spirit Checklist.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lam, L.L., Pavlakis, N., Shitara, K. et al. INTEGRATE II: randomised phase III controlled trials of regorafenib containing regimens versus standard of care in refractory Advanced Gastro-Oesophageal Cancer (AGOC): a study by the Australasian Gastro-Intestinal Trials Group (AGITG). BMC Cancer 23, 180 (2023). https://doi.org/10.1186/s12885-023-10642-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-10642-7