
Abstract
Background
Urothelial carcinoma (UC) is the ninth most commonly diagnosed cancer worldwide, with a 3.8/1 male to female ratio. Platinum-based chemotherapy is the first line standard of care for fit patients with advanced UC. However, despite a response rate (RR) for approximately half of patients receiving standard chemotherapy, durable responses are rare (median progression-free progression (PFS) around 8 months). Recently, immune checkpoint inhibitors (ICI) have emerged as new therapeutic options. Among them, Avelumab, an anti-PD-L1 antibody, was assessed in maintenance treatment, demonstrating an overall survival improvement in the JAVELIN Bladder-100 phase III trial. These findings led to its approval as first line maintenance therapy for patients with locally advanced or metastatic UC who have not progressed on prior platinum-containing chemotherapy. However, disease progression as best response was noticed for 37% of patients under Avelumab as maintenance treatment.
UC has targetable genomic alterations, including DNA damage repair (DDR) alterations. DDR deficiency is known to major sensitivity to both platinum-based chemotherapy and PD-1/PD-L1 blockade and the combination of ICI and PARP inhibitors showed promising results. It therefore warrants to assess the interest of combining ICI plus PARP inhibitors as maintenance treatment in UC patients.
Methods
The TALASUR trial is a single-arm multicenter phase 2 study aiming to assess the antitumor activity of the combination of Avelumab with Talazoparib among patients with locally advanced/metastatic UC in maintenance therapy after platinum-based chemotherapy.
The primary objective is to determine the efficacy of the combination, assessed through PFS. Secondary objectives are as follows: safety profile of the association, objective response, duration of tumoral response, disease control rate, time to subsequent therapy, quality of life. A blood and tumor collections will be also constituted.
Patient will receive the combination therapy of daily oral Talazoparib (1 mg/day) and intra-venous Avelumab 800 mg on days 1 and 15, in a 28-day cycle. Fifty patients will be enrolled.
Discussion
Talazoparib with Avelumab combination may have additive activity when administrated jointly. We hypothesize that combination will increase the antitumor activity in UC first line maintenance setting with an acceptable safety profile.
Trial registration
NCT04678362, registered December 21, 2020. Protocol version: Version 1.3 dated from 2020 09 11.
Similar content being viewed by others


Background
Metastatic urothelial carcinoma management
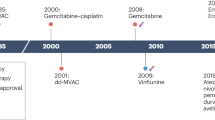
Significant improvements were made in the management of metastatic urothelial carcinoma (mUC) with immune checkpoint inhibitors (ICI). In first line setting, for patients who are deemed ineligible for platinum-based therapy but whose tumor expresses PD-L1, 2 checkpoint inhibitors (Pembrolizumab, Atezolizumab) are currently approved. After platinum-based chemotherapy failure, five molecules anti-PD-1 or PD-L1 antibodies (Pembrolizumab, Atezolizumab, Avelumab, Nivolumab and Durvalumab) have obtained FDA/EMA approval [1, 2]. More recently, the Javelin Bladder 100 phase III trial assessing Avelumab, an anti-PD-L1 antibody, as switch maintenance therapy in mUC patients who achieved stable disease or response after 4–6 cycles of first line platinum-based chemotherapy. The study demonstrated significantly prolonged overall survival (OS) with maintenance Avelumab + Best Supportive Care (BSC) vs BSC alone (median OS = 21.4 versus 14.3 months, p < 0.001) as well as progression-free survival (PFS) (median PFS 3.7, versus 2.0 months, p < 0.001) [3]. The objective response rate (ORR) was 9.7% of patients in the Avelumab + BSC arm versus 1.4% in the BSC alone arm. The safety profile was similar to that of other ICI. Despite these encouraging results showing long term durable ORR, 37% of patients had disease progression as best response. There is thus a need to find alternative/combination approaches for the treatment of mUC.
The emerging role of DNA damage repair and PARP inhibition in UC
The characterization of the genomic landscape of bladder UC has identified targetable genomic alterations, including a high number of loss of function mutations in DNA damage repair (DDR) genes gathered in the concept of “BRCAness phenotype” [4, 5]. The Cancer Genome Atlas analysis of bladder UC found 34% of BRCAness phenotype including CHEK1/2, RAD51, BRCA1/2, ATM, ATR, MDC1, and FANCF gene alterations [4]. Necchi et al. underwent a comprehensive genomic profiling of 479 upper tract UC and 1 984 bladder UC: they found 4.6 and 4.9% of specific BRCA1/2 alterations, respectively [5]. Studies that evaluated the frequency of germline pathogenic variants in the DDR genes found alterations in 10% of cases [6, 7].
The “BRCAness phenotype” has been shown to confer sensitivity to the Poly ADP-Ribose Polymerase (PARP) inhibitors. Several PARP inhibitors (PARPi) are nowadays approved in various tumor types (breast, prostate, pancreas and ovarian carcinoma) [8]. For some tumor types, the benefit is not restricted to BRCA mutation (e.g. ovarian, breast or prostate cancer). PARPi have also demonstrated efficacy in unselected-biomarkers ovarian carcinoma, in maintenance setting after response to platinum-based chemotherapy.
The prevalence of these somatic mutations in homologous recombination (HR) pathway as well as their association with improved platinum sensitivity suggests PARP inhibition to be a rational target for the treatment of mUC, but the biomarker selection remains unclear.
PARPi were assessed in monotherapy or in association in UC patients, molecularly selected or not. The ATLAS trial failed to demonstrate the efficacy of Rucaparib, a PARPi as single agent treatment in patients with previously treated locally advanced/unresectable or metastatic UC, in a biomarker-unselected population [9]. Similarly, the Meet-Uro12 trial, a randomised phase II study, failed to demonstrate the efficacy of niraparib monotherapy plus BSC versus BSC alone as maintenance treatment for patients with unresectable or metastatic biomarker-unselected UC whose disease did not progress after first line platinum-based chemotherapy [10, 11]. In this study, BRCAness phenotype was retrospectively evaluated: HRR alterations were found in 44.7% of patients, illustrating platinum sensitivity of mUC with DDR deficiency. In contrast, the Atlantis trial is a biomarker-directed, multi-arm phase II study, in maintenance setting for mUC after platinum-based chemotherapy response or stable disease [12, 13]. Results from the Atlantis rucaparib arm in DDR-selected mUC were recently presented: maintenance rucaparib was associated with promising benefit as compared to placebo (median PFS 35 versus 15 weeks respectively, HR 0.53, 80% CI 0.30–092; p = 0.07). Similarly, the BISCAY trial is an open-label, biomarker-directed, multi-arm phase Ib study in mUC after platinum-based chemotherapy failure [14]. DDR deficiency was found in 14% of tumors and interestingly, 54% of these tumors were considered tumor mutation burden (TMB) high. The combination of olaparib, a PARPi and durvalumab, an anti-PD-L1 inhibitor, demonstrated modest but interesting clinical benefit, with an ORR of 35.7% in this biomarker-selected population.
Maintaining a tumoral response and prolonging PFS are clinically meaningful purposes for clinical trials exploring novel agents in advanced UC. The objectives of a maintenance strategy are to increase and/or prolong the benefit of chemotherapy using drugs which safety profile allowing their long term use.
Rationale for combining Talazoparib and Avelumab
PARPi and ICI are good candidates thanks to their acceptable safety profile, and synergistic activity. They are already approved in this maintenance strategy. Preclinical and clinical data suggest that PARPi may have additive activity when administrated in combination with ICI, through different mechanisms, independently of the DNA damage repair status [15, 16]: PARPi-mediated unrepaired DNA damages indeed modulate the tumor immune microenvironment which might promote responsiveness to ICIs.
The Neodurvarib trial assessed Durvalumab in combination with Olaparib in UC neo-adjuvant setting. It highlighted promising results, with complete pathological response for 44.5% of patients and acceptable toxicity [17].
Talazoparib is an orally bioavailable potent inhibitor of PARP [18]. It selectively binds to PARP and prevents PARP-mediated DNA repair of single strand DNA breaks via the base-excision repair pathway. This enhances the accumulation of DNA strand breaks, promotes genomic instability and eventually leads to apoptosis. In a first-in-man phase 1 study, Talazoparib showed clinical activity in various solid tumors. It is already approved for the treatment of metastatic breast cancer patients with an inherited BRCA mutation [19, 20].
The phase Ib/II trial assessing the association of Avelumab and Talazoparib (Javelin PARP Medley trial—NCT03330405) in locally advanced or metastatic solid tumors, is ongoing [21]. Results from the phase Ib step indicate an acceptable safety profile of the combination. The RP2D (recommended phase 2 dose) was Avelumab 800 mg per intravenous route every two weeks (Q2W) and Talazoparib 1 mg per os daily given (0.75 mg daily in case of moderate renal impairment). Results from phase II biomarker-selected advanced breast cancer cohorts (22 and 23 patients) were also presented. Beyond the acceptable safety profile, the combination demonstrated meaningful antitumor activity, with 18% and 30% ORR according to the cohorts and responses generally durable. The 3-arm phase III trial Javelin Ovarian PARP100 was designed to evaluate the efficacy and safety of avelumab in combination with chemotherapy followed by maintenance therapy of avelumab plus talazoparib in treatment-naïve patients with locally advanced or metastatic ovarian cancer. The trial was stopped prematurely for futility (insufficient efficacy)of avelumab monotherapy in this setting but did not showed safety concerns about the association [22].
Purpose
We propose to investigate the efficacy and the safety of the association Talazoparib and Avelumab as maintenance therapy in platinum-sensitive metastatic or locally advanced UC. Patients will be selected according to platinum sensitivity, to allow excluding poor prognosis tumors and potentially enriching the proportion of patients whose tumor harbours alterations in genes involved in DNA damage repair. Predictive interest of DDR biomarkers will be assessed as part of ancillary analyses.
Methods / design
We propose a one-arm single-stage multicenter phase II study aiming to assess the interest of combining Talazoparib and Avelumab as maintenance therapy in platinum-sensitive metastatic or locally advanced UC. The TALASUR protocol and this manuscript have been written in accordance with standard protocol items, namely recommendations for interventional trials (SPIRIT).
Primary outcome
The primary objective of this phase II study is to determine the efficacy of a maintenance treatment combining Talazoparib with Avelumab in patients with locally advanced/metastatic UC with a stable disease or objective response to a platinum-based chemotherapy.
Efficacy will be measured by the PFS defined as the time from initiation of Talazoparib plus Avelumab treatment to the first documented disease progression per RECIST 1.1 criteria (Response Evaluation Criteria in solid Tumors) based on investigator’s assessment, or death due to any cause, whichever occurs first.
Secondary outcomes
The secondary objectives are to evaluate:
-
▪The safety profile of Talazoparib plus Avelumab according to NCI CTCAE v5.0 criteria
-
▪The efficacy of the combination, for whole study population, and according to the response to the initial chemotherapy, through:
-
• Overall Survival, defined as the time from Talazoparib + Avelumab initiation to death whatever cause
-
• ORR, defined as the proportion of patients with partial or complete response, according to RECIST 1.1 criteria
-
• ORR, defined as the proportion of patients with partial or complete response, according to RECIST 1.1 criteria
-
• Duration of tumoral response, defined as the time elapsed between first date of objective response (complete or partial response) and date of first documented disease progression according to RECIST 1.1 criteria or date of death, due to any cause, whichever occurs first
-
• Disease control rate, defined as the proportion of patients with stable disease, partial or complete response, according to RECIST 1.1 criteria
-
• Duration of treatment by Talazoparib plus Avelumab taken together and separately defined as the time elapsed between the first dose of treatment and the date of permanent discontinuation of Talazoparib and/or Avelumab regardless of cause (Patients still on treatment with any study drug at the time of database lock will be censored).
-
• Time to subsequent therapy (TST), defined as the time elapsed between the date of initiation of Talazoparib plus Avelumab and the date of initiation of subsequent systemic therapy, respectively (Patients who did not receive any subsequent treatment or who have died prior to receiving subsequent treatment will be censored).
-
-
▪To assess Quality of life (QoL) of patients under Talazoparib plus Avelumab treatment with:
-
•Scores of quality of life assessed through the French version of the self-administered standardized questionnaire EORTC QLQ-C30 and EuroQoL EQ-5D.
-
•Survival without deterioration in QoL as assessed using the QLQ-C30 questionnaire.
-
-
▪To constitute a blood and tumor banking for ancillary biological explorations.
These translational researches may include the determination of the patients’ and disease’s characteristics of tumors (PD-L1 status, microsatellite instability (MMR), tumor mutation burden (TMB), specific molecular alterations of the homologous recombination pathway from genetic material derived from tumor tissue or circulating tumoral DNA). The relationship between clinical and molecular (genomic, metabolic, and/or proteomic) markers and the safety profile, as well as the efficacy of the combination Avelumab plus Talazoparib may also be considered.
Study population
Eligibility criteria are precised in Table 1. The TALASUR study addresses patients with locally advanced/metastatic UC with a stable disease or objective response to a platinum-based chemotherapy.
Study sites
The list of study sites is indicated on https://clinicaltrials.gov/ct2/show/NCT04678362. The participation of 12 French Centres highly involved in the GETUG intergroup is planned (Table 2).
Study treatments and procedures
The study schedule is resumed in Fig. 1 and an overview of study assessments and procedures is presented in Table 3.

TALASUR study schedule
Eligible patients must be enrolled by medical oncologists within 8 weeks after the last dose of chemotherapy and start the combined treatment within 28 days after inclusion. Patients will receive a treatment by Talazoparib plus Avelumab as follows:
-
Talazoparib will be administered at the daily dose of 1 mg given orally in a 28-day cycle.
In case of mild renal impairment (creatinine clearance between 30-59 mL/min) at inclusion, patients will receive the daily dose of 0.75 mg.
-
Avelumab will be administered by intravenous (I.V.) route over 60 min at the dose of 800 mg on D1 and D15, in a 28-day cycle.
For Avelumab treatment, patients have to be premedicated with an antihistamine and with paracetamol 30 to 60 min prior to the first 4 infusions. If the fourth infusion is completed without an infusion-related reaction, premedication for subsequent doses should be administered at the discretion of the physician. No premedication is required for Talazoparib.
Any toxicity observed during the course of the study could be managed by discontinuation of Talazoparib or dose reduction based on severity and clinical presentation. A maximum of three dose reductions are allowed for Talazoparib: it is recommended to first reduce to 0.75 mg daily, then to 0.5 mg daily, and lastly to 0.25 mg.daily. As for Avelumab, there is no dose adaptation: dosing delay or discontinuation may be required based on individual safety and tolerability.
Concomitant treatments must be prescribed in accordance with the Avelumab and Talazoparib and collected in the e-CRF.
The combined treatment will be continued until disease progression, unacceptable toxicity or discontinuation for any cause (patient or investigator consideration).
The patient’s participation in the study will be stopped once the treatment with both study drugs will be discontinued regardless of cause. A period of 8 weeks with no doses of Avelumab and/or Talazoparib is deemed to be treatment discontinuation and withdrawal of the patient from the study. Subsequent care will be given outside of the context of the TALASUR study.
In case of complete response under study drugs, the combined treatment will be stopped after 2 years after the documentation of this complete response.
Tumor evaluation (thoracic and abdomino-pelvic) will be performed on CT-scan or MRI machine at baseline, every 8 weeks, at the end of study treatment, and thereafter every 8 weeks in the absence of tumoral progression. Disease assessment evaluation will be determined locally by the investigator according to RECIST criteria 1.1.
A Quality of life assessment (EORTC QLQ-C30 and EQ-5D self-administered questionnaires) will be performed at baseline, every 8 weeks and at the end of treatment.
Statistical design overview
The TALASUR study is a multicenter single-arm phase II trial.
Sample size calculation
Based on JAVELIN Bladder 100 results (PFS of 3.4 months in Avelumab arm), the following hypotheses were chosen: a median PFS of 4 months will be considered as an unacceptable efficacy endpoint whereas a median PFS of 7 months will be considered as an acceptable endpoint showing efficacy of this combination. Assuming a 5% (one tailed) type I error, according to a one-arm survival single stage design, 45 assessable patients will be required in the trial to ensure a 80% power (SWOG Cancer Research and Biostatistics). The median PFS should be longer than 5.9 months for the study to be positive. To anticipate 10% of non-assessable patients, we plan to enrol 50 patients overall over 18 months of inclusion.
Statistical analyses
All outcomes will be analysed based on intention-to-treat principle. PFS, OS and local/distant disease control rates will be estimated via the Kaplan–Meier method and median follow-up will be calculated using the reverse Kaplan–Meier method, from which the median and 95% CI will be calculated. Patients who will die without reported prior progression will be considered to have progressed on the day of their death. Patients without documented death at the time of the final analysis will be censored at the date of the last follow-up that verified absence of progression. As for the exploratory analyses, we will consider using proportional Cox regression to evaluate the association between time-to-event outcomes and covariates such as PD-L1 status.
Adverse events during the study will be tabulated overall and further tabulation will be made based on time of occurrence and relationship to treatment. The latter excludes events unrelated or not likely related to treatment, but includes events for which the relationship with treatment is not assessable. Tolerability will be summarized by duration of treatment, reasons of discontinuation, dose reduction rates and reasons for dose reductions.
Quality of life (QoL) scores and changes from baseline scores will be described for selected primary scales. Missing values will be calculated such that if at least half the items from the scale will be completed, it will be assumed that the missing items will have values equal to the average of those items present. The Z-test or the non-parametric Wilcoxon–Mann–Whitney tests will be used to evaluate the evolution in global health status and other dimensions of the EORTC QLQ-C30 and EQ-5D. Survival without deterioration in QoL will be defined as the time interval between the date of initiation of Talazoparib plus Avelumab treatment and date of the first deterioration. Deterioration from baseline is defined as a “minimal clinically important difference” (MCID) of > 10-point out of 100. Deterioration will be considered in at least one of the following dimensions: global health-related QoL, and fatigue, without significant clinical improvement subsequently or death, regardless of cause. Survival without deterioration in QoL will be estimated with the Q-TWIST (Quality-Adjusted Time Without Symptoms or Toxicity) method.and.
All the analyses will be performed in R (Version 4.0.2) and will be two sided with p value less than 0.05 considered as significant.
Independent data monitoring committee
An Independent Data Monitoring Committee (IDMC) will be set-up to ensure the protection of patients, to ensure the ethical conduct of the study, to evaluate the benefit/risk ratio of the study and to insure an independent review of the scientific outcomes during and at completion of the study.
The committee will include a biostatistician, a pharmacologist and a medical oncologist.
The members of the IDMC will be consulted at least before the trial initiation, and at the final analysis.
Data management
A Web Based Data Capture (WBDC) system will be used for data collection and query handling. The investigator will ensure that data are recorded on the eCRFs as specified in the study protocol and in accordance with the instructions provided.
The investigator ensures the accuracy, completeness, and timeliness o
f the data recorded and of the provision of answers to data queries according to the Clinical Study Agreement. The investigator will sign the completed eCRFs. A copy of the completed eCRFs will be archived at the study site.
Treatment discontinuation and withdrawal from study
The reasons for why a patient may discontinue to participate to the study or interrupt study treatment include the following circumstances.
Treatment discontinuation
Avelumab and/or Talazoparib will be administered until the investigator considers that the patient no longer obtains benefit from it. Avelumab and/or Talazoparib will be interrupted at any time under the following circumstances:
-
Unacceptable toxicity requiring discontinuation of both treatments.
-
A period of 8 weeks with no doses of Avelumab and/or Talazoparib is deemed to be treatment discontinuation and withdrawal of the patient from the study.
-
Disease progression
-
Need to initiate another systemic anti-tumor treatment
-
Patient’s decision (the data already collected during the search can be kept and exploited unless the patient opposes it)
-
Patient withdrawal
-
Investigator’s decision
-
Concomitant illness or other reason that necessitates stopping treatment of the study
-
Protocol violation
-
Patient lost to follow-up
-
Death
-
Study terminated by Sponsor
In the event of study termination due to disease progression, non-response to treatment, or early termination of the study for any cause, management of patient including subsequent anti-tumor treatment will be at the investigator’s discretion.
Withdrawal from study
Reasons a patient may discontinue treatment include, but are not limited to:
-
Treatment failure/confirmed disease progression
-
Major protocol violation
-
Intolerable toxicity
-
Concomitant disease or other reason requiring the discontinuation of treatment
-
Patient request (withdrawal of consent for further treatment)
-
Investigator’s request (with detailed documentation of reasoning)
-
Non-compliance of patient
-
Trial termination by the sponsor
-
Pregnancy
-
Death
Discussion
The recommended first line treatment of mUC is a platinum-based chemotherapy. This treatment is efficient with an ORR higher than > 50%. However, the PFS remains short (around 7.7 months) and the chemotherapy is too toxic to be used in a prolonged time. Traditionally, maintenance systemic therapy refers to the utilization of regimens with less toxicity given to patients whose cancer is controlled after the initial upfront treatment. The aim of maintenance treatment is to prolong the disease control obtained with initial treatment. This concept has already been shown with PARPi in ovarian carcinoma, as well as, more recently, with Durvalumab in lung carcinoma and Avelumab in UC which is now the standard of care of maintenance treatment after response or stability to the first line platinum treatment.
The prevalence of somatic mutations in HR genes in UC, as well as the known relationship between DDR deficiency and platinum sensitivity, and the recent results from the Atlantis trial with rucaparib treatment in DDR tumors suggest Talazoparib, a PARPi, to be a target for a maintenance treatment of UC. Moreover, there is a strong rationale with both pre-clinical and clinical data to associate Avelumab and Talazoparib.
In addition to the assessment of the clinical outcomes of Avelumab plus Talazoparib in mUC in maintenance therapy after platinum-based chemotherapy, the translational explorations will shed light on some potential determinants of the efficacy of the dual treatment. UC is indeed known to be a heterogeneous disease characterized by genomic instability and a high mutation rate. According to the recent findings from the exploratory biomarkers analysis of the Javelin 100 trial, OS appeared to be modestly associated with mutations in individual genes such as BRCA1 and BRCA2. However, considering molecular signatures, an OS benefit in PD-L1 positive subgroup and tumors with mutated DDR genes was observed, reinforcing the relevance of investigating the molecular markers that may impact on the benefit of a maintenance treatment combining Avelumab and Talazoparib [23].
Conclusion
Findings from recent phase II trials showed some clinical benefits of PARPi treatment in UC, but the tumor biomarker selection and the treatment indication remain debated. The single arm phase II TALASUR trial will evaluate the association of Avelumab and Talazoparib in maintenance setting in platinum sensible and unselected biomarker mUC to determine the efficacy and the safety of the combination in this indication and investigate predictive biomarkers in explorative ancillary studies.
Availability of data and materials
Not applicable.
Abbreviations
- BSC:
-
Best supportive care
- CR:
-
Complete response
- CT:
-
Computed Tomography
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- DCIS:
-
Ductal carcinoma in situ
- ECOG:
-
Eastern Cooperative Oncology Group
- FDA:
-
Food and Drug Administration
- FFPE:
-
Formalin-fixed, paraffin-embedded
- GETUG:
-
Groupe d’Etude des Tumeurs Uro-Génitales
- ICI:
-
Immune checkpoint inhibitor
- IDMC:
-
Independent Data Monitoring Committee
- MRI:
-
Magnetic Resonance Imaging
- NCI:
-
National Cancer Institute
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PARPi:
-
Poly(ADP-ribose) polymerase inhibitor
- PD 1:
-
Programmed death 1
- PDL 1:
-
Programmed death ligand 1
- PFS:
-
Progression-free survival
- PR:
-
Partial response
- QoL:
-
Quality of life
- Q-TWIST:
-
Quality-Adjusted Time Without Symptoms or Toxicity
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- RR:
-
Response rate
- SPIRIT:
-
Standard Protocol Items, Recommendations for Interventional Trials
- TST:
-
Time to subsequent therapy
- UC:
-
Urothelial carcinoma
References
Witjes JA, Bruins HM, Cathomas R, Compérat EM, Cowan NC, Gakis G, et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur Urol. 2021;79:82–104.
Flaig TW. NCCN Guidelines Updates: Management of Muscle-Invasive Bladder Cancer. J Natl Compr Cancer Netw JNCCN. 2019;17(5.5):591–3.
Powles T, Park SH, Voog E, Caserta C, Valderrama BP, Gurney H, et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. 2020;383(13):1218–30. https://doi.org/10.1056/NEJMoa2002788.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22.
Necchi A, Madison R, Pal SK, Ross JS, Agarwal N, Sonpavde G, et al. Comprehensive Genomic Profiling of Upper-tract and Bladder Urothelial Carcinoma. Eur Urol Focus. 2020. https://doi.org/10.1016/j.euf.2020.08.001.
Nassar AH, AbouAlaiwi S, AlDubayan SH, Moore N, Mouw KW, Kwiatkowski DJ, et al. Prevalence of pathogenic germline cancer risk variants in high-risk urothelial carcinoma. Genet Med Off J Am Coll Med Genet. 2020;22:709–18.
Carlo MI, Zhang L, Mandelker D, Vijai J, Cipolla CK, Robson ME, et al. Cancer predisposing germline mutations in patients (pts) with urothelial cancer (UC) of the renal pelvis (R-P), ureter (U) and bladder (B). J Clin Oncol. 2017;35(15_suppl):4510–4510.
Schettini F, Giudici F, Bernocchi O, Sirico M, Corona SP, Giuliano M, et al. Poly (ADP-ribose) polymerase inhibitors in solid tumours: Systematic review and meta-analysis. Eur J Cancer Oxf Engl. 1990;2021(149):134–52.
Grivas P, Loriot Y, Morales-Barrera R, Teo MY, Zakharia Y, Feyerabend S, et al. Efficacy and safety of rucaparib in previously treated, locally advanced or metastatic urothelial carcinoma from a phase 2, open-label trial (ATLAS). BMC Cancer. 2021;21:593.
Vignani F, Hamzaj A, Giorgi UD, Tambaro R, Basso U, Gamba T, et al. 800TiP Meet-URO 12: A randomized phase II trial of niraparib versus best supportive care (BSC) as maintenance treatment in patients with locally advanced or metastatic urothelial cancer (UC) whose disease did not progress after completion of first-line platinum-based chemotherapy. Ann Oncol. 2020;31:S607.
Vignani F, Tambaro R, De Giorgi U, Giannatempo P, Bimbatti D, Carella C, et al. J Clin Oncol. 2022;40(6_suppl):442–442.
Fulton B, Jones R, Powles T, Crabb S, Paul J, Birtle A, et al. ATLANTIS: a randomised multi-arm phase II biomarker-directed umbrella screening trial of maintenance targeted therapy after chemotherapy in patients with advanced or metastatic urothelial cancer. Trials. 2020;21:344.
Crabb SJ, Hussain SA, Soulis E, Hinsley S, Dempsey L, Trevethan A, et al. A randomized, double blind, biomarker selected, phase II clinical trial of maintenance PARP inhibition following chemotherapy for metastatic urothelial carcinoma (mUC): Final analysis of the ATLANTIS rucaparib arm. J Clin Oncol. 2022;40(6_suppl):436–436.
Powles T, Carroll D, Chowdhury S, Gravis G, Joly F, Carles J, et al. An adaptive, biomarker-directed platform study of durvalumab in combination with targeted therapies in advanced urothelial cancer. Nat Med. 2021;27:793–801.
Teo MY, Bambury RM, Zabor EC, Jordan E, Al-Ahmadie H, Boyd ME, et al. DNA Damage Response and Repair Gene Alterations Are Associated with Improved Survival in Patients with Platinum-Treated Advanced Urothelial Carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23:3610–8.
Peyraud F, Italiano A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers. 2020;12:1502.
Rodriguez-Moreno JF, de Velasco G, Bravo Fernandez I, Alvarez-Fernandez C, Fernandez R, Vazquez-Estevez S, et al. Impact of the combination of durvalumab (MEDI4736) plus olaparib (AZD2281) administered prior to surgery in the molecular profile of resectable urothelial bladder cancer: NEODURVARIB Trial. J Clin Oncol. 2020;38(6_suppl):542–542.
Hoy SM. Talazoparib: First Global Approval. Drugs. 2018;78:1939–46.
de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017;7:620–9.
Litton JK, Hurvitz SA, Mina LA, Rugo HS, Lee K-H, Gonçalves A, et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: final overall survival results from the EMBRACA trial. Ann Oncol Off J Eur Soc Med Oncol. 2020;31:1526–35.
Yap TA, Konstantinopoulos P, Telli ML, Saraykar S, Beck JT, Galsky MD, et al. Abstract P1–19–03: JAVELIN PARP Medley, a phase 1b/2 study of avelumab plus talazoparib: Results from advanced breast cancer cohorts. Cancer Res. 2020;80(4 Supplement):P1-19-03-P1-19–03.
Eskander RN, Ledermann JA, Birrer MJ, Fujiwara K, Gaillard S, Richardson GE, et al. JAVELIN ovarian PARP 100 study design: Phase III trial of avelumab + chemotherapy followed by avelumab + talazoparib maintenance in previously untreated epithelial ovarian cancer. J Clin Oncol. 2019;37(8_suppl):TPS9.
Powles T, Sridhar SS, Loriot Y, Bellmunt J, Mu XJ, Ching KA, et al. Avelumab maintenance in advanced urothelial carcinoma: biomarker analysis of the phase 3 JAVELIN Bladder 100 trial. Nat Med. 2021. https://doi.org/10.1038/s41591-021-01579-0.
Acknowledgements
We acknowledge the GETUG (Groupe d’Etude des Tumeurs Uro-Génitales) for its support in scientific collaboration. We thank the Data Processing Centre (DPC) of the North West Canceropole (Centre de Traitement des Données du Cancéropôle Nord-Ouest) in charge of data management. We also thank the members of the Independent Data Monitoring Committee. We are grateful to all the patients who will consent to participate.
Scientific approval
This trial was scientifically approved by the GETUG Intergroup (academic clinical research group specializing in uro-genital tumors / Groupe d’Etude des Tumeurs Uro-Génitales).
Funding
This trial (NCT04678362) is granted by Pfizer SAS that also provides Talazoparib and Avelumab for participating patients. Pfizer SAS is not involved in the design and conduct of the study, nor in the collection, management, analysis, and interpretation of the data. It is not involved in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
EC, BC, JL and ATV wrote the manuscript and devised the study concept and design. JL was responsible for overseeing the statistical section. EC, BC, JL ATV and FJ have been involved in drafting the manuscript or revising it critically for important intellectual content. EC, BC and ATV supervised the entire work. All authors (EC, BC, JL, PEB, ZN, EM, IB, MC, NG, JB, RT, FJ, JMG, and ATV) have given their final approval of the version to be published. Each author has been sufficiently involved in the work to take public responsibility for appropriate portions of the content.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study has received ethical approval from the Comité de Protection des Personnes EST IV in December 2020 (N° EudraCT: 2020–001105-24) and from National Agency for Medical and Health products Safety (Reference: MEDAECNAT-2020–08-00012_2020-001105–24) in October 2020. All patients will be proposed to participate by the medical oncologists who will give them an information file; all patients will give their written informed consent before any study-related assessment start.
Consent for publication
Not applicable.
Competing interests
This trial (NCT04678362) is granted by Pfizer SAS that also provides Talazoparib and Avelumab for participating patients.
None of the authors has a conflict of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Coquan, E., Clarisse, B., Lequesne, J. et al. TALASUR trial: a single arm phase II trial assessing efficacy and safety of TALazoparib and Avelumab as maintenance therapy in platinum-Sensitive metastatic or locally advanced URothelial carcinoma. BMC Cancer 22, 1213 (2022). https://doi.org/10.1186/s12885-022-10216-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-10216-z