
Abstract
Background
The aim of the study was to enhance colorectal cancer prognostication by integrating single nucleotide polymorphism (SNP) and gene expression (GE) microarrays for genomic and transcriptional alteration detection; genes with concurrent gains and losses were used to develop a prognostic signature.
Methods
The discovery dataset comprised 32 Taiwanese colorectal cancer patients, of which 31 were assayed for GE and copy number variations (CNVs) with Illumina Human HT-12 BeadChip v4.0 and Omni 25 BeadChip v1.1. Concurrent gains and losses were declared if coherent manners were observed between GE and SNP arrays. Concurrent genes were also identified in The Cancer Genome Atlas Project (TCGA) as the secondary discovery dataset (n = 345).
Results
The “universal” concurrent genes, which were the combination of z-transformed correlation coefficients, contained 4022 genes. Candidate genes were evaluated within each of the 10 public domain microarray datasets, and 1655 (2000 probe sets) were prognostic in at least one study. Consensus across all datasets was used to build a risk predictive model, while distinct relapse-free/overall survival patterns between defined risk groups were observed among four out of five training datasets. The predictive accuracy of recurrence, metastasis, or death was between 61 and 86% (cross-validation area under the receiver operating characteristic (ROC) curve: 0.548-0.833) from five independent validation studies.
Conclusion
The colorectal cancer concurrent gene signature is prognostic in terms of recurrence, metastasis, or mortality among 1746 patients. Genes with coherent patterns between genomic and transcriptional contexts are more likely to provide prognostication for colorectal cancer.
Similar content being viewed by others
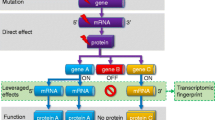


Background
Colorectal cancer (CRC) is the leading cause of human malignancies in Taiwan and ranks third among all cancer deaths [1]. CRC is also a molecularly heterogeneous disease; microarray and reserve transcription-polymerase chain reaction (RT–PCR) experiments have revealed a number of molecular subtypes based on gene expression (GE) profiles, with some displaying associations with disease prognosis or treatment response [2,3,4,5,6,7,8,9,10,11]. For instance, Oncotype DX (Genomic Health Inc., Redwood City, CA) was developed as a 12-gene recurrence score. Other signatures included the 18-gene ColoPrint (Agendia Inc., Irvine, CA) and ColoGuide Ex, which is a 13-gene signature using an Affymetrix (Thermo Fisher Scientific, Waltham, MA) exon-based microarray. At the same time, CRC also shows chromosomal instability, with largely unknown clinical significance [12, 13].
The precise etiology of sporadic CRC remains undetermined, as opposed to hereditary familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC, ref. [14]). Chromosomal instability might be one of the critical initiatives of sporadic CRC. Cancers can result from progressive accumulation of genetic aberrations with amplified regions containing oncogenes and deleterious regions with tumor suppressor genes. Additionally, cytogenetic analyses have identified oncogenes and tumor suppressors at breakpoints of recurrent chromosomal aberrations [15, 16]. In addition, genomic aberrations could impact GE by complex transcriptional regulation, and genes displaying coherent patterns between the genome and transcriptome are hypothesized to serve as potential biomarkers for prognostication [17, 18].
We used two high-throughput technologies, single nucleotide polymorphism (SNP) and GE microarrays, to conduct an integrated study unraveling critical genes with prognostication in CRC. Although a number of GE signatures have been proposed [2,3,4,5,6,7,8,9,10,11], there are unsettled concerns regarding reproducibility and clinical applicability in conjunction with conventional pathological factors [19]. A more sophisticated methodology must be established before molecular signatures can be widely adopted in clinical practice. Herein, we presented a novel GE signature for CRC based on concurrent genes.
Methods
The study protocol was reviewed and approved by the institutional review board (IRB) of Cathay General Hospital. Written informed consent was obtained from all the participants after explanation by the investigators (MHS and CCH). An overview of the study design is depicted in Fig. 1.

Overview of study design (CRC: colorectal cancer, GE: gene expression, SNP: single nucleotide polymorphism, b/w: between, TCGA-COAD: The Cancer Genome Atlas colon adenocarcinoma, RFS: relapse-free survival, OS: overall survival, GSE: Gene Expression Omnibus series accession number)
CRC samples
CRC samples were prospectively and consecutively collected during surgery. Enrollment criteria included CRC patients who had never received neoadjuvant therapy, were in clinical stages 0 (in situ) to III (no systemic spread) and had no concurrent secondary malignancy. Enrolled patients were managed according to standard guidelines with regular follow-up. All patients with resectable CRC were treated with curative surgeries.
The cancerous tissues were snap frozen and stored in liquid nitrogen below − 80 °C with RNAlater reagent (Qiagen, Germantown, MD) to stabilize RNA. The frozen samples were dissected into slices of 1-2 mm thickness, and more than 90% cancerous content was a prerequisite for microarray experiments.
Microarray experiments: GE arrays
Total RNA was extracted from frozen specimens using TRIzol reagent (Invitrogen, Carlsbad, CA). Purification of RNA was performed using a RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The minimal RNA concentration was set to 100 ng/μl (25 μl) per sample. RNA integration was checked by gel electrophoresis with 2 bands of 18S and 28S indicating satisfactory RNA quality, and an RIN value > 8.0 was pursued to remove heavily degraded samples. Illumina Human HT-12 BeadChip v4.0 (Illumina, San Diego, CA) was used for GE experiments, which provided genome-wide transcriptional coverage of well-characterized genes. Each array on HumanHT-12 v4.0 Expression BeadChip targeted more than 47,000 probes derived from NCBI Reference Sequence (RefSeq) Release 38 (November 7, 2009) and other sources. GE data were normalized using Illumina BeadStudio software Gene Expression Module, and the generated final report files were exported for further analyses, including the AVGSignal spreadsheet.
Microarray experiments: SNP arrays
DNA was extracted from cancerous tissues using a QIAamp DNA mini kit (Qiagen, Valencia, CA) from the same subject undergoing GE experiments. A minimum of 4 μg DNA was required. A Bioanalyzer 2100 (Agilent, Santa Clara, CA) was used to verify the purity and concentration of cancerous DNA with quality control indicated by OD260/280 > 1.8. Illumina Human Omni 25 BeadChip v1.1 was used for SNP array experiments, which featured approximately 2.5 million markers that captured variants down to a minor allele frequency (MAF) of 2.5% and delivered whole genomic coverage across diverse populations. Structural variations, mainly copy number variants (CNVs), were detected. Illumina HiScan array scanners supported genotyping, CNV, and GE profiling. Projects created with BeadStudio were exported, with three spreadsheets, namely, Genotype, Intensity, and BAlleleFrequency, reported separately.
CNV detection
CNV detection began with segmentation of normalized data (Intensity spreadsheet from BeadStudio), followed by identification of common (recurrent) gains and losses across multiple SNP arrays. Circular binary segmentation (CBS) was used to identify regions in each chromosome such that copy numbers in each region were equal [20, 21]. The significance level for the test to accept change points was set to 0.01, and the number of permutations was 1000. The Smoothing and MergeLevels algorithm were applied to enhance efficiency [22, 23]. Based on segmented log ratios, the copy number at a particular genomic location was determined using the median absolute deviation (MAD) of log ratios of each array. High-level CNV (amplification and homozygous deletion) was assigned to regions with segmentation mean log ratios > 1 and < − 1 timed the MAD of each corresponding array. The thresholds for low-level CNV (both gains and losses) were 0.5 and − 0.5 MAD, respectively. Pathway enrichment analyses were based on the BioCarta (URL: https://maayanlab.cloud/Harmonizome/dataset/Biocarta+Pathways) database, evaluating the association between a pathway and regions of gain/loss with an empirical P-value by 1000 times random sampling.
Regions of recurrent CNV within a cohort of samples were identified using the Genomic Identification of Significant Targets in Cancer (GISTIC, ref. [24]). A null distribution of G scores was generated based on 10,000 resamplings. The significance of CNV at a particular genomic location was determined based on a statistical test obtained from the segmentation log ratios of assayed samples. All bioinformatics analyses of CNV were conducted with the CGH Tools v1.3, part of the BRB-ArrayTools [21]. Results of pathway enrichment and GISTIC analyses were reported for CRC cases assayed for SNP arrays.
Concurrent gains and losses
Concurrent gains and losses were detected from common genes across SNP and GE microarrays by using HUGO gene symbols as identifiers. The process of mapping between SNP and GE microarray platforms was performed with the SOURCE (URL: https://source-search.princeton.edu/) or Clone/Gene ID Converter (URL: https://cran.r-project.org/web/packages/IDConverter/index.html), depending on which method provided the greatest number of reliable conversions. For probe reduction, multiple probes/probe sets were reduced to one per gene symbol by using the most variable probe/probe set measured by IQR across arrays.
We integrated GE and CNV data to identify genes whose transcriptional abundance was impacted by CNV. A Gene-centric table was outputted by the CGH Tools and detailed the average log-intensity ratio (calculated from all markers within a gene) rather than the discrete CNV status per gene, which was also used to deduce a value corresponding to each gene for each array in the array-covered genomic regions. This value was used to calculate correlations between CNV and GE arrays to distinguish concurrent genes. Concurrent gains and losses were declared if significant changes in a coherent manner were observed for both GE and SNP microarrays (assessed by Spearman correlation coefficients with P values < 0.05).
Concurrent gene signature and classification algorithms
Concurrent genes were identified from Taiwanese CRC and The Cancer Genome Atlas (TCGA) data. The TCGA-COAD (colon adenocarcinoma) Project level 3 dataset of 345 patients was assayed for both GE and CNV profiles using Agilent 4502A (Agilent, Santa Clara, CA) and Affymetrix Genome-wide SNP 6.0 (Thermo Fisher Scientific, Waltham, MA) microarrays. Clinical, CNV, and GE data were downloaded under the synapse ID syn1461155 as the secondary discovery dataset from the URL (URL: https://www.synapse.org/, ref. [25]). Gene mapping was performed as described in Method E, with an additional source of NetAffy (URL: https://www.affymetrix.com/analysis/netaffx_analysis_center_retired.html).
Rather than identifying common genes from both discovery datasets, the “universal” concurrent gene set was derived statistically. Fisher’s z transformation was used to combine correlation estimates from concurrent genes identified from Taiwanese populations and those from the TCGA-COAD dataset with the mathematical formula as follows:
The combined correlations from independent samples were:
where z is Fisher’s z-transformation, r is the sample correlation, V is variance, and n is the sample size. The universal concurrent gene set was filtered with the predefined threshold of a 10−3 α level and was used for downstream prognostic model construction. SAS/STAT software version 15.1 (SAS Institute Inc., Cary, NC) with the CORR procedure was used for the estimation of z-transformed correlation coefficients.
Microarray datasets
Publicly available microarray datasets were retrieved and fulfilled the purpose of training and validation of the risk predictive model. The primary outcomes were relapse-free or overall survival, and the secondary outcomes were adverse events following curative therapy, such as recurrence, metastasis, or mortality (all were dichotomous outcomes without survival data). Datasets that met the outcome variables were retrieved and are detailed in Supplementary Table 1 (GSE12945, GSE14333, GSE17538, GSE39582, and TCGA_COAD with survival data) and Supplementary Table 2 (GSE5206, GSE9348, GSE18088, GSE18105, and GSE64857 with dichotomous outcomes but without survival data).
Risk predictive model
A CRC risk predictive model for relapse-free/overall survival was constructed using supervised principal component regression [26]. Concurrent genes were first filtered by the univariate Cox proportional hazards regression, and significant genes within a stringent α level of 0.001 were further used to synthesize the first principal component (supergene), which was subsequently used in risk prediction. A continuous prognostic index score was calculated based on the first principal component for each subject within a dataset, and the high- and low-risk groups were defined by the 50th percentile prognostic index score (noninformative prior). A sensitivity analysis was performed with the prognostic index score cutoff between the predicted high- and low-risk group determined by the lowest censored percentage across all studies (the 75th percentile, Supplementary Table 6).
For dichotomous outcomes such as recurrence, metastasis or death, differentially expressed concurrent genes were identified using the univariate two-sample t test at a 0.001 significance level. A global multivariate permutation test (α level of 10− 3) was further used to control false positivity. Multiple methods, including compound covariate predictor, diagonal linear discriminative analysis, 3 nearest neighbors, nearest centroid, and support vector machine (SVM, with default penalty of LIBSVM, ref. [27]), were used to evaluate the prediction accuracy of the CRC risk model (class prediction functions of the BRB- ArrayTools, ref. [21]). For all class prediction methods, leave-one-out cross-validation (LOOCV) was used to calculate the misclassification rate with a permutation P-value reported. For each random permutation of class labels, the entire cross-validation procedure was repeated to calculate the cross-validated misclassification rate with the final P value determined from the proportion of the random permutations giving the least misclassification rate. A minimum of 1000 permutations was required.
As distinct statistic/bioinformatics tools were adopted with different underlying hypotheses and corresponding scenarios, there was no uniform alpha-level across all these tests. Consequently, default alpha-level of each test from the BRB-ArrayTools was followed. Usually reduced α levels were required for multiple testing (Bonferroni correction). Model training and LOOCV were performed within each study, and consensual genes across all microarray datasets were used to build the CRC risk predictive model. Genes were median-centered first within each dataset to avoid introducing bias from extremely high intensities as well as batch effects.
Results
Taiwanese CRC cohort
A total of 88 CRC patients were recruited during the study period between October 2013 and May 2016. There were 51 males and 37 females, with a median age of 63 (range: 33-88) years. There were 81 adenocarcinomas, 2 mucinous adenocarcinomas, and 5 in situ lesions. The anatomical distributions were ascending (21), transverse (9), descending (10), sigmoid (19), rectosigmoid (9), anorectum (18), and overlapping lesions (2). There were 46 low-, 32 intermediate-, and 7 high-grade cases. During the follow-up period of up to 4 years, there were 7 recurrences and 14 metastases.
CNV of 32 Taiwanese CRC patients
The number of unique markers delivered by the Illumina SNP array was 2,267,360, and the frequencies of CNV among 32 Taiwanese CRC patients (31 also assayed for GE) are displayed in Fig. 2. Supplementary Fig. 1 summarizes gain and loss calls on the genome detected on each array. Pathway enrichment analysis with BioCarta showed that there was one pathway enriched in genes with gain and two pathways enriched in genes with loss (Supplementary Table 3, ref. [28]). Frequent CNVs (gain regions) identified by GISTIC are detailed in Supplementary Table 4, including 13q12.2-q12.3 (PDX1, ATP5EP2, CDX2, PRHOXNB, FLT3, LOC100288730, PAN3, and FLT1) and 17q12-q21.2 (NEUROD2, PPP1R1B, STARD3, TCAP, PNMT, PGAP3, ERBB2, C17orf37, GRB7, IKZF3, ZPBP2, GSDMB, ORMDL3, and LOC728129).

Frequency plot of CNV among 32 Taiwanese CRC patients (CNV: copy number variation, CRC: colorectal cancer)
Consensus of universal concurrent genes
The number of concurrent genes was 1582 (P < 0.01) and 2974 (P < 0.01) from the Taiwanese (n = 31 for CRC patients assayed for both platforms) and TCGA-COAD (n = 345) cohorts, respectively, resulting in a common concurrent gene set of 307 genes. The universal concurrent genes, which were synthesized from the combined correlation coefficients with z-transformation, contained 4022 genes at the P < 0.001 level. These candidate genes were evaluated within each of the 10 microarray datasets, and 1655 (2000 probe sets) were filtered as being prognostic in at least one study (Table 1). The consensual genes across studies were used to build a CRC risk predictive model with a significance level determined by a nominal univariate test at 0.01 α level. Candidate genes decreased from 1365 to 1 when the number of agreeing datasets increased from 1 to 5 (Table 2). Finally, a subset of 49 consensual concurrent genes were incorporated into the CRC prognostic model, which was determined by cross-validation. A complete list of the 49 constitutional genes is detailed in Supplementary Table 5.
Survival analysis
A supervised principal component encompassing 49 consensual concurrent genes was used for survival analysis, with the threshold of the 50th percentile of the prognostic index score for risk group construction with LOOCV. Figure 3A to E show relapse-free/overall survival patterns from the GSE12945, GSE14333, GSE17538, TCGA_COAD, and GSE39582 datasets. Except for GSE12945 (log-rank test: P = 0.82), survival discrepancies were observed in 4 out of 5 studies between defined high- and low-risk groups (log-rank test: P < 0.0001, P = 0.0059, P = 0.049 and P = 0.0361, respectively). In order to evaluate the impact of the 50th percentile thresholding, Table 3 summarizes area under the curve (AUC) from time-dependent receiver operating characteristic (ROC) curve, as well as censored/uncensored number from each study. The highest AUC, 0.824, was reported from TCGA_COAD cohort; it should be noticed that it was also this cohort contributed much more samples during the discovery of concurrent genes. A differential bias toward favorable prognostic power for this cohort should be considered.

A to E Relapse-free/overall survival analysis from microarray datasets of GSE12945 (A top left), GSE14333 (B top right), GSE17538 (C middle left), TCGA_COAD (D middle right), and GSE39582 (E bottom) with leave-one-out cross-validation. The high−/low-risk group was defined by the 50th percentile of the prognostic index score determined by the supervised principal component composed of 49 concurrent genes. All survival times were measured in months, except for 3D, which was measured in days
To further evaluate the impact of different thresholds on risk grouping, a sensitivity analysis was performed with a different cutoff of the 75th percentile. Supplementary Table 6 shows the number of predicted high−/low-risk patients and censored cases within each risk group while survival patterns and AUC are detailed in Supplementary Fig. 2. It deserved noticed that with an informatic prior of one-quarter of the assayed samples with the highest prognostic index score being high-risk, discerning ability of the purposed signature was much compromised, and only two out of the five studies with follow-up data showed a significant log-rank test (Supplementary Table 6). These two studies, GSE14333 and GSE 17538, were with a proportion of censored cases most similar to the pre-selected 75th percentile cutoff and reported an optimistic survival advantage among CRC patients predicted into the low-risk group (Supplementary Fig. 2B and C).
Predictive accuracy of CRC risk model
Table 4 shows the predictive accuracy of the concurrent gene-based CRC prognostic model with multiple methods predicting adverse events of recurrence, metastasis, or death during the follow-up period. In general, the Bayesian compound covariate model delivered the best predictive accuracy, with a cross-validation AUC reaching 0.833.
Discussion
CRC is a major gastrointestinal malignancy, while its development and progression involves a complex process with multiple genetic changes. Therefore, deciphering the molecular heterogeneity of CRC will contribute to accurate risk assessment and identify effective therapies. Mainstays of CRC management include early detection by screening (fecal occult blood testing), complete surgical resection of the lesion with regional lymph node dissection, and adjuvant systemic therapy based on the diagnostic stage. Adjuvant therapy, usually with the form of cytotoxic chemotherapy, is mainly determined by the pathological staging system including depth of tumor invasion, regional node involvement and distant metastatic status. Usually stage I CRC is managed with regular surveillance, stage III is deemed with adjuvant therapy; while for stage II disease, there remains a lack of prognostic biomarkers for risk assessment except dMMR (mismatch repair deficiency) status [29].
Beyond anatomical staging, GE patterns of CRC may provide additional prognostication, which is complementary to pathological features. This biological staging will not be achievable without a transcriptome-based molecular profiling. The current study evaluated the prognostic value of concurrent gene sets specific for CRC, and a risk predictive model was proposed. We developed an analytical approach to identify genes with coherent patterns between transcriptome and CNV profiles using matched GE and SNP microarray data to reduce false discoveries in concurrent gene signatures.
CRC is heterogeneous in terms of molecular aberrations, and oncogenesis could originate from chromosomal CNV and manifest as transcriptional alterations. However, relationships between DNA structural variations and mRNA abundance are not always linear, and complex regulatory mechanisms have rarely been addressed, further highlighting the necessity of identifying genes underpinning CRC tumorigenesis. GE studies using microarray, RT–PCR, or digital RNA counting have been advocated and performed widely to search biomarkers for cancer prognostic prediction [2,3,4,5,6,7,8,9,10,11, 17, 18]. Fresh frozen samples of newly diagnosed CRC patients were assayed in an integrated approach to synthesize the purposed concurrent gene signature, which could be used to improve CRC risk stratification, further augmenting treatment outcomes.
The 32 Taiwanese CRC patients assayed with SNP microarrays showed that the most frequent CNVs by GISTIC were 13q12.2-q12.3, and 17q12-q21.2. SNP array-based CNV analysis, which is a molecular cytogenetic method, can detect abnormalities in the number of copies of segments of tumor DNA, with losses or gains from assayed samples indicated from spots showing aberrant intensity signal ratios. Whole-genome SNP arrays can provide insight into the fundamental process of chromosomal instability leading to CRC oncogenesis [30].
Chromosomal aberrations seem to play a major role in regulating transcription [31, 32]. Since genomic imbalance would have a substantial impact on GE, the interplay between CNV and certain GE patterns for sporadic CRC might shed light on underlying molecular processes and the discovery of cancer-related prognostic genes. The main hypothesis underpinning the concurrent gene signature is that cancer may be bred at the chromosomal level with CNV and modulate subsequent GE profiles. Concurrent genes, which were designated to those displaying coherent patterns between tumor genomic and transcriptional alterations, were the filtered candidates for prognostic signature synthesis.
With the prevalence of high-throughput GE studies, hundreds of thousands of genes were measured in a single experiment, and gene filtering became inevitable to derive a clinically applicable signature from high-dimensional GE data [33]. In the current study, concurrent genes were the selection criteria for biomarker discovery and classifier development to identify potential candidates through algorithms integrating SNP and GE microarrays. Both CNV and GE data were available in 31 subjects with Spearman’s correlation coefficients calculated for each gene, resulting in 1582 concurrent genes (P < 0.01). The secondary discovery cohort, comprising 345 patients from TCGA-COAD project, revealed 2974 concurrent genes using the same algorithm (P < 0.01). Instead of using the 307 intersecting genes, we combined correlations from two independent cohorts using z-transformation, and 4022 genes were filtered (P < 0.001). Calling pipelines were the same for both discovery cohorts once the GE abundance and CNV were summarized from a gene-by gene basis, despite different microarray platforms were adopted within each study. The z-transformed correlation coefficients were used to identify the universal concurrent genes in an effort to overcome the unbalanced sample size between two discovery cohorts. This sophisticated statistical framework addressed the sample size discrepancy between two discovery cohorts without losing generalizability.
Publicly available CRC microarray studies were gathered from the NCBI’s Gene Expression Omnibus. Studies reporting relapse-free survival or overall survival, as well as those with dichotomous prognostic outcomes, were included. Processed data were downloaded and analyzed without further modification except for gene centering, since prognostic comparisons were performed within each dataset to avoid batch effects across microarray studies. The 4022 candidate concurrent genes were evaluated within each of the 10 microarray datasets, and 1655 (2000 probe sets) were recognized as being prognostic in at least one study. The final prognostic model was the consensus across 10 microarray datasets. Distinct relapse-free or overall survival patterns were evident from four out of five datasets, and the predictive accuracy of adverse events was between 61 and 86% from another five independent studies. It deserves notice that GE-based predicted risk is always continuous, that’s why an arbitrary threshold for dichotomous stratification is needed for prognostication. The 50th percentile was selected as an uninformative prior, which meant that there was equal chance of being censored/event during survival analysis. With high variability of censored cases across studies (68% ~ 92%), our sensitivity analyses showed that an uninformative prior set to the 50th percentile of the prognostic index score might be a better choice to enhance generalizability for real-world practice. The built-in multiple-methods of the BRB-ArrayTools were adopted for class prediction. For high-dimensional GE data, there is no gold standard for which is the best method for class prediction, so we conducted exhaustive bioinformatics approaches to identify the best model.
As pointed out by Marshall et al. from a review article regarding multi-omics, after 10 years’ progress of tumor mutational and transcriptional profiling in CRC, the prognostic power of modern genetic testing brings only modest benefits in terms of treatment guidance, i.e., who will benefit from adjuvant chemotherapy and if so, what is the optimal duration or intensity? [34]. Indeed, there remains an unmet need of precise risk estimation for proper management of CRC patients while our study provided a plausible multi-gene expression signature for such task. Survival rate of stage II CRC is around 60 to 85% while 25% of patients of this stage will relapse, and once relapse happened, their survival rate will drop drastically. Consequently, guiding the decision of adjuvant cytotoxic chemotherapy for CRC is one of the major priorities for the application of multi-gene expression-based testing.
Therefore, we developed the prognostic model for CRC as an initiative toward personalized and precision medicine.
The biological relevance of the 49 constitutional genes was deciphered. KDM6B (JMJD3) is an epigenetic gene coding for a histone demethylase and is also a VDR co-target that partially mediates the effects of 1,25-(OH)2D3 on the human colon [35]. ATAD5 mediates the cellular response to DNA damage [36]. HIP1R has been harvested from sera of CRC but not from normal blood donors [37]. PBK is a serine/threonine kinase, and its expression is elevated in breast cancer, prostate cancer, and CRC [38]. PBK/TOPK interacts with the DBD domain of the tumor suppressor p53 and modulates the expression of transcriptional targets, including p21 [39]. PIAS2 regulates the IFN-gamma signaling pathway, affecting tumor development in non-small-cell lung cancer [40]. FLNA, DUSP14, and FAS are implicated in the MAPK pathway [41]. FAS is relevant to p53 and the apoptosis pathway [42], and THBS1 interacts with TGF-beta in glioblastoma [43]. POLD2 is involved in DNA replication and mismatch/base excision repair [44]. Finally, CSTF1 participates in mRNA polyadenylation [45]. In addition, many signature genes remained undetermined regarding their roles in CRC pathogenesis, while our integrated analysis was an initial step toward understanding their relevance in CRC initiation and progression.
There were some limitations of the study. First, as sequencing was not conducted, it was not possible to evaluate the impact of tumor DNA sequence variants upon GE; consequently, genes impacted by both mutations and CNV were not selected for signature construction. Future prospective validation studies using retrospective formalin-fixed paraffin-embedded (FFPE) samples are warranted to show the true prognostic value of concurrent gene signatures. The benefits of FFPE samples include readily available pathological archives, affordable quantitative RT–PCR testing or digital RNA counting rather than much more expensive microarrays, and an abundance of clinical information from retrospective cohorts. We hope the integrated approach could lead to the discovery of potential biomarkers with prognostic value for CRC to determine the most efficient adjuvant therapy based on risk stratification, especially for stage II patients [46]. With prognostic validation of the concurrent gene signature, those predicted into the high-risk group should be managed with postoperative adjuvant therapy to reduce their risk of recurrence and metastasis and should be assayed by targeted sequencing of actionable mutations such as KRAS and BRAF [47]. On the other hand, stage II CRC patients categorized into the low-risk group by concurrent gene signature may avoid toxic chemotherapy under regular postoperative surveillance.
Conclusions
The concurrent gene risk predictive model is prognostic for CRC recurrence, metastasis, or mortality as well as relapse-free/overall survival from 1746 patients. CRC oncogenesis might originate from tumor CNV and manifest through transcription as GE profiles. Genes with coherent patterns between chromosomal and transcriptional contexts are more likely to serve as potential biomarkers for sporadic CRC. With prognostic validation of the concurrent gene signature, more precise risk assessment will be achieved to overcome the molecular heterogeneity of CRC, and the results will provide further opportunities for personalized therapy.
Availability of data and materials
Processed gene expression values and a gene-centric table pertaining the average log-intensity ratio per gene from comparative genomic hybridization experiments (SNP microarrays), as well as sample information are detailed in the Additional file 3. Raw microarray data of Taiwanese CRC is deposited to the NCBI GEO with the accession number GSE197802.
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- GE:
-
Gene expression
- CNV:
-
Copy number variation
- TCGA:
-
The Cancer Genome Atlas project
- ROC:
-
Receiver operating characteristic
- CRC:
-
Colorectal cancer
- RT–PCR:
-
Reserve transcription-polymerase chain reaction
- FAP:
-
Familial adenomatous polyposis
- IRB:
-
Institutional review board
- MAF:
-
Minor allele frequency
- CBS:
-
Circular binary segmentation
- MAD:
-
Median absolute deviation
- GISTIC:
-
Genomic identification of significant targets in cancer
- COAD:
-
Colon adenocarcinoma
- SVM:
-
Support vector machine
- FFPE:
-
Formalin-fixed paraffin-embedded
References
Health Promotion Administration, Ministry of Health and Welfare, the Executive Yuan. Cancer Registry Annual Report, Republic of China. 2015. http://www.hpa.gov.tw/BHPNet/Web/HealthTopic/TopicArticle.aspx?No=201312230005&parentid=200712250030. Accessed 24 June 2018.
Kelley RK, Venook AP. Prognostic and predictive markers in stage II colon cancer: is there a role for gene expression profiling? Clin Colorectal Cancer. 2011;10:73–80.
Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol. 2011;29:4611–9.
Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol. 2013;31:4512–9.
Kennedy RD, Bylesjo M, Kerr P, et al. Development and independent validation of a prognostic assay for stage II colon cancer using formalin- fixed paraffin-embedded tissue. J Clin Oncol. 2011;29:4620–6.
Allen WL, Jithesh PV, Oliver GR, et al. The colorectal cancer disease-specific transcriptome may facilitate the discovery of more biologically and clinically relevant information. BMC Cancer. 2010;10:687.
Niedzwiecki D, Frankel W, Venook AP, et al. Association between Col Dx assay result and recurrence-free interval in stage II colon cancer patients on CALGB (Alliance) 9581. J Clin Oncol. 2014;32(suppl 3):455a.
Salazar R, Roepman P, Capella G, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29:17–24.
Salazar R, Capdevila J, Rosenberg R, et al. Comparison of Colo Print risk classification with clinical risk in the prospective PARSC trial. J Clin Oncol. 2014;32(suppl 3):465a.
Agesen TH, Sveen A, Merok MA, et al. Colo Guide Ex: a robust gene classifier specific for stage II colorectal cancer prognosis. Gut. 2012;61:1560–7.
Sveen A, Agesen TH, Nesbakken A, et al. Colo Guide Pro: a prognostic 7-gene expression signature for stage III colorectal cancer patients. Clin Cancer Res. 2012;18:6001–10.
Ried T, Meijer GA, Harrison DJ, Grech G, Franch-Expósito S, Briffa R, et al. The landscape of genomic copy number alterations in colorectal cancer and their consequences on gene expression levels and disease outcome. Mol Asp Med. 2019;69:48–61.
Oliveira DM, Santamaria G, Laudanna C, Migliozzi S, Zoppoli P, Quist M, et al. Identification of copy number alterations in colon cancer from analysis of amplicon-based next generation sequencing data. Oncotarget. 2018;9(29):20409–25.
Medina Pabón MA, Babiker HM. A review of hereditary colorectal cancers. [Updated 2020 Nov 19]. In: StatPearls. Treasure Island: StatPearls Publishing; 2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538195/.
Pollack JR, Perou CM, Alizadeh AA, et al. Genome-wide analysis of DNA copy number changes using cDNA microarrays. Nat Genet. 1999;23:41–6.
Oostlander AE, Meijer GA, Ylstra B. Microarray-based comparative genomic hybridization and its applications in human genetics. Clin Genet. 2004;66:488–95.
Huang CS, Lu TP, Liu CY, Huang CJ, Chiu JH, Chen YJ, et al. Residual risk stratification of Taiwanese breast cancers following curative therapies with the extended concurrent genes signature. Breast Cancer Res Treat. 2021;186(2):475–85.
Huang CC, Tu SH, Lien HH, Jeng JY, Huang CS, Huang CJ, et al. Concurrent gene signatures for han chinese breast cancers. PLoS One. 2013;8(10):e76421.
Johnston PG. Identification of clinically relevant molecular subtypes in colorectal cancer: the dawning of a new era. Oncologist. 2014;19(5):568–73.
Olshen AB, Venkatraman ES, Lucito R, et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–72.
Simon R, Lam A, Li MC, et al. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007;3:11–7.
Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–63.
Willenbrock H, Fridlyand J. A comparison study: applying segmentation to array CGH data for downstream analyses. Bioinformatics. 2005;21:4084–91.
Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–12.
Omberg L, Ellrott K, Yuan Y, Kandoth C, Wong C, Kellen MR, et al. Enabling transparent and collaborative computational analysis of 12 tumor types within The Cancer Genome Atlas. Nat Genet. 2013;45(10):1121–6.
Bair E, Tibshirani R. Semisupervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2:511–22.
Chang CC, Lin CJ. LIBSVM: a library for support vector machines. ACM TIST. 2011;2:1–27.
Simon R, Wang SJ. Use of genomic signatures in therapeutics development in oncology and other diseases. Pharmacogenomics J. 2006;6(3):166–73.
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349(3):247–57.
Hieronymus H, Murali R, Tin A, Yadav K, Abida W, Moller H, et al. Tumor copy number alteration burden is a pancancer prognostic factor associated with recurrence and death. Elife. 2018;7:e37294.
Shlien A, Malkin D. Copy number variations and cancer susceptibility. Curr Opin Oncol. 2010;22(1):55–63.
Shao X, Lv N, Liao J, Long J, Xue R, Ai N, et al. Copy number variation is highly correlated with differential gene expression: a pancancer study. BMC Med Genet. 2019;20(1):175.
Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J Natl Cancer Inst. 2007;99(2):147–57.
Marshall JL, Peshkin BN, Yoshino T, et al. The essentials of multiomics. Oncologist. 2022;27(4):272–84.
Pereira F, Barbáchano A, Silva J, Bonilla F, Campbell MJ, Muñoz A, et al. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum Mol Genet. 2011;20(23):4655–65.
Giovannini S, Weller MC, Hanzlíková H, Shiota T, Takeda S, Jiricny J. ATAD5 deficiency alters DNA damage metabolism and sensitizes cells to PARP inhibition. Nucleic Acids Res. 2020;48(9):4928–39.
Scanlan MJ, Welt S, Gordon CM, Chen YT, Gure AO, Stockert E, et al. Cancer-related serological recognition of human colon cancer: identification of potential diagnostic and immunotherapeutic targets. Cancer Res. 2002;62(14):4041–7.
Dou X, Wei J, Sun A, Shao G, Childress C, Yang W, et al. PBK/TOPK mediates geranylgeranylation signaling for breast cancer cell proliferation. Cancer Cell Int. 2015;15:27.
Hu F, Gartenhaus RB, Eichberg D, Liu Z, Fang HB, Rapoport AP. PBK/TOPK interacts with the DBD domain of tumor suppressor p53 and modulates expression of transcriptional targets including p21. Oncogene. 2010;29(40):5464–74.
Zhao YC, Tang D, Yang S, Liu H, Luo S, Stinchcombe TE, et al. Novel variants of ELP2 and PIAS1 in the interferon gamma signaling pathway are associated with non-small-cell lung cancer survival. Cancer Epidemiol Biomark Prev. 2020;29(8):1679–88.
Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford). 2016;2016:baw100.
Li Y, Raffo AJ, Drew L, Mao Y, Tran A, Petrylak DP, et al. Fas-mediated apoptosis is dependent on wild-type p53 status in human cancer cells expressing a temperature-sensitive p53 mutant alanine-143. Cancer Res. 2003;63(7):1527–33.
Daubon T, Léon C, Clarke K, Andrique L, Salabert L, Darbo E, et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat Commun. 2019;10(1):1146.
Zheng Y, Sheng S, Wang H, Jia X, Hu Y, Qian Y, et al. Identification of Pold2 as a novel interaction partner of protein inhibitor of activated STAT2. Int J Mol Med. 2012;30(4):884–8.
Neve J, Patel R, Wang Z, Louey A, Furger AM. Cleavage and polyadenylation: ending the message expands gene regulation. RNA Biol. 2017;14(7):865–90.
Varghese A. Chemotherapy for stage II colon cancer. Clin Colon Rectal Surg. 2015;28(4):256–61.
D'Haene N, Fontanges Q, De Nève N, et al. Clinical application of targeted next-generation sequencing for colorectal cancer patients: a multicentric Belgian experience. Oncotarget. 2018;9(29):20761–8.
Acknowledgments
This work contains materials presented orally at the 49th Annual Scientific Meeting of the Taiwan Surgical Association, which was held in Taipei, Taiwan during March 20, 2016.
Funding
This study was sponsored in part by the Ministry of Science and Technology, Taiwan with grant number NSC102-2314-B-281-003-MY3 and Cathay General Hospital with grant number MA-10312.
Author information
Authors and Affiliations
Contributions
MHS drafted the manuscript, CJH conducted microarray experiments, CYL performed pathological examinations, TFH and YYS participated in subject enrollment and panel discussion, CSH and CCH initiated the study and approved the final submitted manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study protocol was reviewed and approved by the institutional review board (IRB) of Cathay General Hospital. Written informed consent was obtained from all the participants after explanation by the investigators (MHS and CCH). All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
All authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Microarray datasets with survival outcomes (n = 1331). Supplementary Table 2. Microarray datasets with dichotomous outcomes pertaining to adverse events (n = 415). Supplementary Table 3. Pathways enrichment in genes with gain and loss. Supplementary Table 4. Gain and loss regions and associated genes by GISTIC among 32 Taiwanese CRC patients (GISTIC: Genomic Identification of Significant Targets in Cancer, CRC: colorectal cancer). Supplementary Table 5. Complete list of the concurrent gene signature. Supplementary Table 6. Summary of censored case numbers in high- and low-risk groups and area under the curve (AUC) from time-dependent receiver operating characteristic (ROC) curve from survival analysis (gene only model, RFS: relapse-free survival, OS: Overall survival, CRC: colorectal cancer, GEO: gene expression omnibus) with risk-group thresholding set to the 75th percentile.
Additional file 2: Supplementary Figure 1.
Gain-loss plot of the genome of 32 Taiwanese CRC SNP microarrays (CRC: colorectal cancer SNP: single nucleotide polymorphism). Supplemental Figure 2. A-E Relapse-free/overall survival analysis from microarray datasets of GSE12945 (2A, top left), GSE14333 (2B, top right), GSE17538 (2C, middle left), TCGA_COAD (2D, middle right), and GSE39582 (2E, bottom) with leave-one-out cross-validation. The high−/low-risk group was defined by the 75th percentile of the prognostic index score determined by the supervised principal component composed of 49 concurrent genes. All survival times were measured in months, except for 3D, which was measured in days.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, MH., Huang, CJ., Ho, TF. et al. Colorectal cancer concurrent gene signature based on coherent patterns between genomic and transcriptional alterations. BMC Cancer 22, 590 (2022). https://doi.org/10.1186/s12885-022-09627-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-09627-9