
Abstract
Background
Pancreatic ductal adenocarcinoma (PDAC) is the major subtype of pancreatic cancer and head PDACs show distinct characteristics from body/tail PDACs. With limited studies based on Asian population, the mutational landscape of Asian PDAC remains unclear.
Methods
One hundred fifty-one Chinese patients with head PDAC were selected and underwent targeted 425-gene sequencing. Genomic alterations, tumor mutational burden, and microsatellite instability were analyzed and compared with a TCGA cohort.
Results
The genomic landscape of Chinese and Western head PDAC had identical frequently-mutated genes including KRAS, TP53, SMAD4, and CDKN2A. KRAS hotspot in both cohorts was codon 12 but Chinese PDACs containing more G12V but fewer G12R variants. Potentially pathogenic fusions, CHD2-BRAF and KANK1-MET were identified in two KRAS wild-type patients. Serum cancer antigens CA125 and CA19-9 were positively associated with SMAD4 alterations while high CEA was enriched in wild-type CDKN2A subgroup. The probability of vascular invasion was lower in patients with RNF43 alterations. The nomogram developed including histology grade, the mutation status of SMAD4, TGFBR2, and PREX2 could calculate the risk score of prognoses validated by Chinese and TCGA cohort.
Conclusions
Chinese head PDAC contained more KRAS G12V mutation than Western population. The well-performed nomogram may improve post-operation care in real-world practice.
Similar content being viewed by others


Background
Pancreatic cancer is one of the most deadly cancers with a five-year survival rate of less than 9% [1]. The prognosis of pancreatic patients is highly associated with the diagnosis stage. For early-stage patients, the five-year survival rate can reach 37% but for those with distant metastases, it drops to 3%. The incidence rate of pancreatic cancer is trending upward in China and worldwide [1, 2]. Pancreatic ductal adenocarcinoma (PDAC) is the leading histological subtype and covers over 90% of all pancreatic cancers [3]. 80% of PDACs are located at the head of the pancreas, which arises from different embryonic origins compared with the tail of the pancreas. The ventral bud forms the posterior part of the head or uncinate process, while the dorsal bud forms the rest of the pancreas [4]. The prognosis between head PDAC and body/tail PDAC were extremely controversial. Some studies indicated primary head PDAC has a better prognosis than body/tail PDAC [5, 6], while others suggested the opposite outcome in resectable or early advanced PDAC [7, 8]. As the comprehensive studies revealed 4 molecular subtypes of PDAC, squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine (ADEX), the squamous subtype was associated with a poor prognosis [9, 10]. Later studies found head and body/tail PDAC have different gene expression signatures and are rich in different molecular subtypes, with body/tail PDAC containing more squamous subtype and less immunogenic subtype than head tumors, which may contribute to their diverse clinical manifestations and outcomes [11, 12].
Previous whole-genome studies have investigated the mutational profiles of PDAC in Western populations and uncovered four abundantly common mutations, including KRAS, TP53, SMAD4, and CDKN2A [13, 14]. Due to the limited studies based on the Asian population, the genetic landscape of Asian PDAC patients remains unclear. Several genes were found survival-related in PDAC [15, 16]. However, no risk stratification was developed based on intra-tumor genetic heterogeneity.
The incidence of mismatch repair deficiency in PDAC is only 2% according to a Danish cohort study [17]. But it’s still worth investigating further as they may benefit from immune therapy especially under the circumstance of very limited treatment options for PDAC patients. Tumor biomarker CA19-9 is applied in clinical practice for PDAC diagnosis, treatment guidance, and follow-ups [18]. Serum tumor marker carcinoembryonic antigen (CEA) and carbohydrate antigen CA125 are widely used in colorectal can ovarian cancer screening. Over the past few years, it is revealed they may benefit PDAC diagnosis and postoperative monitor [19, 20]. However, the correlation between these antigen levels and genomic alterations is rarely studied. To comprehensively study the genetic alterations in the Chinese PDAC patients and if these alterations affect the clinical outcome, 151 Chinese PDAC patients were retrospectively investigated and a nomogram was established to calculate postoperative risk score to predict prognosis.
Methods
Patient cohort and samples
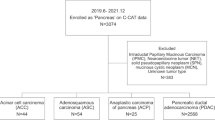
A total of 153 patients diagnosed with head PDAC and underwent surgery (both resectable and palliative) in the First Affiliated Hospital of Nanjing Medical University (also known as Jiangsu Province Hospital), China between October 2017 to February 2019 were retrospectively assessed in this study in accordance with the Declaration of Helsinki. The study was approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University(2020-SR-273), and informedconsents were obtained from all participants. Patients who received neoadjuvant chemotherapy or died of post-operation complications within 30 days after surgery were excluded. Formalin-fixed, paraffin-embedded (FFPE) tumor samples were obtained from all patients for DNA extraction and sequencing. Two patients were excluded from the following analysis whose samples failed the quality control process.
The western cohort that consisted of 91 head PDAC patients was identified from The Cancer Genome Atlas dataset (version # 2016–01-28). Whole-exome sequencing data of the TCGA cohort were analyzed.
DNA extraction, library preparation, and targeted sequencing
Genomic DNA was extracted from FFPE specimen using QIAamp DNA FFPE Tissue Kit (Qiagen), according to the manufacturers’ protocols. DNA was quantified using the dsDNA HS Assay Kit on a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA). The complete DNA concentrations are listed in Supplementary Table S5 with a median concentration of 116 ng/μL (range: 9.3—368 ng/μL). The extracted DNA was also qualified using a Nanodrop2000 (Thermo Fisher Scientific, Waltham, MA) and the same amount of DNA (2000 ng) was uploaded for the following sequencing process. For the five samples whose total amount of extracted DNA was below 2000 ng, all extracted DNA was used (837—1890 ng) and all had passed the following quality control. Sequencing libraries were prepared using the KAPA Hyper Prep Kit (KAPA Biosystems). Genomic DNA was sheared into 200–350 bp fragments using the Covaris M220 instrument (Covaris) and underwent end-repairing, A-tailing, and ligation with indexed sequencing adapters sequentially. Libraries were then amplified by PCR and purified using Agencourt AMPure XP beads. For targeted enrichment, DNA libraries were pooled for hybridization using customized xGen lockdown probes (Integrated DNA Technologies) for 425 cancer-related genes. Captured libraries were subjected to PCR amplification with KAPA HiFi HotStart ReadyMix (KAPA Biosystems). The purified library was quantified using the KAPA Library Quantification Kit (KAPA Biosystems), and its fragment size distribution was analyzed using a Bioanalyzer 2100. Enriched libraries were amplified and subjected for next-generation sequencing (NGS) on Illumina Hiseq4000 platforms (Illumina) using paired-end sequencing to a targeted mean coverage depth of 700 × , which was controlled by data collection software (Illumina).
Sequencing data processing
FASTQ files were processed with Trimmomatic for quality control. Sequencing data were mapped to the Human Genome version 19 (hg19) using the Burrows-Wheeler Aligner [21]. PCR duplicates were removed by Picard (available at https://broadinstitute.github.io/picard/) and the Genome Analysis Toolkit (GATK) was used to perform local realignments around indels and base quality recalibration [22]. Single nucleotide variants and indels were called by VarScan2 and HaplotypeCaller / UnifiedGenotyper in GATK, with the mutant allele frequency (MAF) cutoff as 0.5% and a minimum of three unique mutant reads. Common SNPs were removed using dbSNP and the 1000 Genome project [23]. The resulting somatic variants were further filtered through an in-house list of recurrent sequencing errors that were generated from over 10,000 normal control samples on the same sequencing platform.
Copy number variations (CNVs) were called as losses or gains relative to the overall sample-wide estimated ploidy as previously described [24, 25]. Arm gain or loss was called when more than 50% of the chromosome have copy number gain or loss. Gene fusion (common fusion regions/introns captured in the target panel) was called using DELLY [26]. Tumor mutational burden (TMB) was defined as the number of somatic synonymous mutations per megabase in each sample, with hotspot/fusion mutations excluded.
Fifty-two microsatellite loci are incidentally captured and evaluated during the targeted 425 gene panels. Based on previous validation studies, a fraction of > 0.4 (> 40% unstable loci) was considered microsatellite unstable [27, 28]. Structural variants were detected using FACTERA (Fusion And Chromosomal Translocation Enumeration and Recovery Algorithm) with the default parameter [29]. Likely germline mutations were identified using a computational prediction method, namely Toseq (Genseeq Technology), which is an algorithm developed using machine learning based on past archived patients’ mutational features.
Data collection and analysis
Clinical pathological features including age, sex, tumor diametre, microscopic vascular and perineural invasion, pTNM stage, resection margin, family, and personal cancer history were collected in this study. Resection margin status was classified into R2 (macroscopically positive), R1 (macroscopically negative but tumor found within less than 1 mm from the margin under the microscope), and R0 (macroscopically and microscopically negative). AJCC staging manual 7th edition was used to normalize tumor stage in Chinese cohort with the TCGA cohort. Tumor stage in other parts of this article was under the guidance of AJCC staging manual 8th edition.
Statistical analysis and nomogram development
Data were analyzed using R 4.0.1 [30]. Categorical variables between groups were compared using χ2 or Fisher’s exact test. Continuous variables between groups were compared using two-sided Mann–Whitney U test should the variables failed to obey normal distribution and using Student’s t-test if they follow normal distribution. Kaplan–Meier method was used to determine median overall survival (OS) and the significance of survival analysis was determined by the log-rank test. A nomogram was developed to predict one-year survival post-operation based on multivariate Cox regression using R package “rms”. Variables with a P value of less than 0.1 in univariable analysis and met proportional hazard assumption were chosen for multivariable Cox regression. Patients in Chinese cohort were assigned in chronological order to two groups, training group and Chinese external validation group for nomogram validation, which contains 92 and 49 patients, respectively. 65 patients in the western cohort with available CNV data comprised the second validation cohort. X-tile was used to determine the cut-off of the risk score calculated by the nomogram [31]. Other R packages used in this study include “ComplexHeatmap”, “ggplot2”, “survival”, “survminer”, “waterfall”, and “Hmisc”. Stage IV cases were excluded in survival analysis. P < 0.05 was considered statistically significant.
Results
Clinical and pathological features
A total of 151 Chinese head PDAC patients were enrolled in this study. The median age at diagnosis was 63 (ranging from 31 to 85) and 50.3% (76/151) were male (Table 1). The majority (80.1%, 121/151) of patients have stage I or II PDAC. 111 patients had lymph nodes metastasis, including regional (105/151, 69.5%), non-regional (3/151, 2.0%), and both (3/151, 2.0%) lymph nodes metastasis. Other pathology stages, histology grade, and invasion status were summarized in Table 1. Additionally, 4 out of 151 (2.6%) patients had solitary liver metastasis and thirteen (8.6%) patients reported cancer history including colorectal (4/13), breast (3/13), endometrial (2/13), bladder(2/13), esophageal (1/13), gastric (1/13), and basal cell skin cancer (1/13). In comparison, the TCGA (n = 91) had a higher proportion of early-stage patients (stage I/II: 95.6%, 87/91). The pathology T stage and histologic grade were significantly higher in Chinese cohort, while N stage was higher in TCGA cohort. More patients in the TCGA cohort achieved microscopically margin-negative resection (53.8% vs. 33.1%). The invasion information was not available from TCGA database.
Genetic landscape of Chinese cohort and comparison with TCGA cohort
The genomic mutation profiles of all 151 patients in Chinese cohort were generated by the panel NGS targeting 425 cancer-related genes. The median TMB was 5.7 Mutations/Mb (ranging from 0 to 73.6). As shown in Fig. 1A, the most frequently mutated genes were KRAS (94.7%), TP53 (81.5%), SMAD4 (33.8%), and CDKN2A (25.2%) which were also at the top in TCGA cohort suggesting a similar mutational pattern in Chinese and Western populations. By comparing the frequencies of commonly mutated genes, DNA damage repair pathway, and other oncogenes between this study and TCGA cohort, the majority of them showed roughly equal frequency such as TGFBR2 (9.3% vs. 6.6%, P-value 0.63) and PREX2 (6.0% vs. 3.3%, P-value 0.54). However, Chinese cohort tended to have more TP53 mutations (81.5% vs. 67.0%) but no DNMT3A alteration (0% vs. 5.5%), the P-value of which were 0.013 and 0.007, respectively (Fig. 1B).

Concurrent mutations in Chinese cohort and incidence comparison with TCGA cohort. A The oncoprint of all patients in the Chinese head PDAC cohort. The top panel includes the most frequently mutated genes. The genes in the middle and the bottom panel are DNA damage repair pathway related and other oncogenic genes, respectively. The alteration frequency of each gene is labled on the left side. B The alteration frequency comparison between the cohort in this study and the TCGA cohort with a coloured scale. The P-value less than 0.05 is statistically significant and highlighted in red. C A bar plot shows the proportion of each KRAS alteration subtype in this study and the TCGA cohort
The correlations between clinicopathological features and genomic mutations were investigated in the Chinses cohort. Serum tumor marker CEA and carbohydrate antigens, CA125 and CA19-9, were measured preoperatively. CA19-9 level didn’t correlate to tumor stage in the Chinese cohort (P = 0.85). A strong correlation between CEA and CDKN2A was observed as CEA level was significantly higher in patients with wild-type CDKN2A (Supplementary Figure S2A, mean 8.37 vs. 4.10, P = 0.038). Furthermore, both preoperative CA125 and CA19-9 levels were positively associated with SMAD4 alterations (P = 0.009 & 0.008, respectively, Supplementary Figure S2B-C), which were enriched in older patients(> 50-year-old) (Supplementary Figure S2D, P = 0.031).
As vascular invasion occurred in nearly half cases (65/151, 43.0%) of Chinese cohort, the association between genomic mutations and vascular invasion were investigated. As shown in Supplementary Figure S3, RNF43 alteration was associated with negative microscopic vascular invasion, the odds ratio of which was 0.18 (95%CI: 0.02–0.84, P-value 0.01).
GNAS mutations were found in all three colloid carcinomas and six conventional PDACs. All three colloid carcinomas and six conventional PDAC have intraductal pancreatic neoplasm(IPMN) adjacent to invasive carcinoma, which was identified in 20(20/151, 13%) cases. Most GNAS mutations(8/9, 89%) were located in codon 201 (R201C, R201H & R201L). 8 patients harboring GNAS alteration also had KRAS mutation. The one without concurrent KRAS mutation had a histological appearance of colloid carcinoma, with somatic ATM and APC mutation. TCGA cohort showed co-occurrent GNAS and RNF43 mutations(P = 0.024), but such a relationship was not found in Chinese cohort(P = 0.20). Survival analysis showed no difference between GNAS/RNF43 altered patients and wild-type patients. RICTOR, PREX2, TGFBR2 mutations were associated with IPMN-associated PDAC(P < 0.05). No correlations were found between other histological subtypes and gene alterations.
KRAS mutation in Chinese and TCGA cohort
As KRAS was mutated in over 90% of patients in both Chinese and TCGA cohort, the alteration subtypes of KRAS mutations were futher investigated. As shown in Fig. 1C, the hotspot of KRAS is codon 12 which contributed to 93.8% and 92.9% of all KRAS mutations in this study and TCGA cohort, respectively. The proportion of KRAS G12D in the two cohorts was similar (this study 40.7% vs. TCGA 48.2%) while Chinese cohort had a higher mutation rate of G12V (43.5% vs. 22.4%) but lower with G12R (9.0% vs. 20.0%) comparing to TCGA. The TCGA cohort also had a unique subtype, G12H(1/91), which wasn’t detected in any patients from Chinese cohort. These G12 mutational subtypes were found not to be associated with OS as analyzed in this study (Supplementary Figure S1A). To be noted, ten stage IV patients were excluded from all survival analyses. Other KRAS subtypes mainly occurred in codon 59 and 61, including single or multiple amino acid substitution, with quite low frequency in both cohorts (Fig. 1C). BRAF missense mutations were identified in two KRAS-mutation patients at low allele frequency, which occurred in BRAF V413M and G469A, respectively.
The mutational status of eight patients with wild-type KRAS in Chinese cohort were examined and results showed that they all harbored other RAS pathway-related gene alterations including BRAF, BRCA1/2, EGFR, MET, TP53, TSC1, and TYMS.
Structural variances in Chinese head PDAC cohort
In Chinese cohort, a total of 35 CNVs were detected in 22 patients involving 19 genes and two-thirds (23/35) were amplification (Fig. 1A). The most frequently amplified gene was DLL3 (n = 5) followed by AKT2 (n = 3), while CDKN2A (n = 4), CDKN2B (n = 3), and SMAD4 (n = 2) incurred copy number loss. Furthermore, 14 patients were detected with gene rearrangements (n = 20). It’s worth noting that two KRAS wild-type patients (P74 and P75) harbored potential pathogenic fusions. P74 had a CHD2-BRAF rearrangement which maintained the intact kinase domain of BRAF encoded by exon 11 to 18 (Supplementary Figure S4A). No gene mutations but a MET gene rearrangement was found in P75 by the targeted panel NGS, where the intact MET kinase domain was fused to the coiled-coils of KANK1 (Supplementary Figure S4B).
Mismatch repair (MMR) deficiency (d-MMR) and microsatellite instability (MSI) events
The reported frequencies of d-MMR in PDAC varied greatly and here in the present study nine patients (9/151, 6%) were found with somatic and/or germline MMR mutations, including MLH1, MSH2, MSH6, PMS1, and POLD1 (Table 2). Only one MSH2 splice mutation was reported likely pathogenic in the database. Others have uncertain significance or weren’t recorded. Among the four patients with germline MMR mutations, two had MSI which was also detected in a third patient (P124) harboring somatic MSH2 mutation and accompanied by high TMB. The three MSI patients in Chinese cohort displayed a better prognosis as no one died of PDAC in 12 to 29 months follow-up comparing to a median survival of 18 months in microsatellite stable patients. However, survival analysis found no significant association between MSI and prolonged survival (Supplementary Figure S1B, P = 0.14). Only four patients (4/151, 2.6%) were reported with a TMB over 20 mutations/Mb which were all identified as d-MMR and three of them harbored germline mutations. The remaining one was the above-mentioned P74. Statistical analysis found no correlations between high TMB and prolonged OS (P-value = 0.43, Supplementary Figure S1C). Meanwhile, all four germline d-MMR patients had previous cancer history and/or first-degree relatives’ cancer history (Table 2).
Nomogram calculating risk score and predicting prognosis
To establish a model to predict the prognosis of PDAC patients, Chinese cohort was divided into training (n = 92) and validation (n = 49) cohorts after excluding 10 stage IV patients. The clinical characteristic distribution of the two sub-cohorts remained comparative (Table 1). Univariate and multivariate analyses were performed on training group to evaluate the association between all factors and OS. As shown in Table 3, the four factors with a P-value of less than 0.1 in the univariate analysis were included in the multivariate analysis: histology grade, SMAD4, TGFBR2, and PREX2 mutations (Fig. 2A). A risk score was calculated based on the nomogram and the cut-off of 15 was determined by X-tile [31]. Patients with a risk score of over 15 were considered as high risk whose one-year mortality probability was over 28%. The median OS of low-risk patients was 23.0 months while for the high-risk group, it dropped to 10.5 months (Fig. 2B). The performance of the nomogram was then assessed in the Chinese validation cohort and TCGA validation cohort. As shown in Fig. 2C, the median OS of low-risk and high-risk patients in the Chinese validation cohort was 29.0 and 16.0 months, respectively with a P-value of 0.0347. Similarly, in the TCGA validaton cohort, which included PDAC in all sites, the nomogram was able to accurately predict the prognosis with a 0.31 HR (95% CI: 0.16–0.58, Fig. 2D).

Nomogram for risk score calculation and its performance validation. A The nomogram calculating the risk score of one-year mortality probability based on the selected four features. The status of each feature corresponds to the score on the top panel. The risk score is the sum of the scores corresponding to each feature which then represents the one-year mortality probability according to the scale bars. The risk score of 15 is the cutoff of the high and low risk groups. The overall survival (OS) curves of patients with high (> 15, red) and low (≤ 15, blue) in the Chinese training cohort (B), Chinese validation cohort (C), and the TCGA cohort (D) are shown. Median OS (MOS), HR (95% CI), and P-value are labeled on the right-up corner of each figure
Discussion
In the present study, a gene-related nomogram was developed to predict 1-year postoperative risk and investigated the genetic landscape of head PDAC based on the Chinese population. Nomogram was internally and externally validated and shows good performance in PDAC at all sites and races. It’s worth pointing out that different technologies were used in the two cohorts for DNA sequencing. Chinese cohort used extracted DNA from FFPE tumor samples which then underwent a panel NGS targeting 425 cancer-related genes. In contrast, the TCGA cohort used fresh-frozen samples and WES for mutational analysis. Other than that, the average examined lymph node number in the TCGA cohort is higher than Chinese cohort (19 vs. 16) which may explain the overall lower pathology N stage observed in Chinese cohort. A higher TP53 mutational frequency (81.5%) was observed in Chinese cohort than the TCGA cohort (67.0%) which is also higher than another pan-site PDAC study (73.5%) reported by Singhi et al. [32]. Besides, more high-TMB (> 20 mutations/Mb) patients were present in Chinese cohort (2.6%) comparing to previous studies, in which the reported TMB-high rates were less than 1% [32, 33]. This might be caused by the relatively high incidence of d-MMR in this study (9%) as other studies showed about 1% d-MMR by NGS and IHC [34]. Furthermore, whether MSI is associated with survival remained inconclusive and no significant association was found between MSI and prolonged survival in Chinese cohort [35]. However, three MSI patients did display a better prognosis who were all alive at the latest follow-up in the period of 12, 19, and 29 months compared to the median OS of 18 months in microsatellite stable patients. And these MSI patients were all TMB-high, which is consistent with previous observations [36].
Studies have found that somatic GNAS and RNF43 mutations were recurrently identified in IPMN [37, 38], so the relationship between GNAS/RNF43 mutations and PDAC with adjuvant IPMN was investigated. Majority of the GNAS mutations occurred in codon 201, which is consistent with the previous report [38]. Studies had shown colloid carcinomas of the pancreas were arose in association with IPMN, and GNAS codon 201 mutations can be identified in the majority of colloid carcinoma [39, 40], which are also verified in present study. RNF43 alteration wasn’t related to any histological subtypes or co-altered with GNAS, however, alterations of RNF43 may contribute to negative vascular invasion. In human hepatocellular carcinoma (HHC), RNF43 overexpression frequently occurred and study had shown correlated with RNF43 expression and vascular invasion [41].
PDAC is a tumor driven by KRAS mutation, which explained its striking prevalence of over 94% [42]. Studies have shown KRAS downstream signaling is affected by different KRAS mutations in an allele-specific manner [43, 44]. Therefore, personalized therapies according to specific KRAS mutations are being extensively investigated. Several inhibitors have been developed targeting KRAS G12C mutation to inhibit KRAS signaling, which unfortunately is quite rare in PDAC, and not present in Chinese cohort [45, 46]. Other inhibitors targeting KRAS G12V, G12D, or G12A were currently under pre-clinical development and showed promising results in pancreatic patient-derived cell lines and xenografts [47]. The present study revealed a difference in Chinese and Western KRAS mutation spectrum. Chinese PDAC had a higher mutation rate of KRAS G12V but a lower KRAS G12R mutation rate comparing to TCGA cohort. Two recent studies presented at the 2020 ESMO Congress about Chinese PDAC molecular profiling also reported a similar KRAS G12 spectrum as this study [48, 49].
Our study showed Chinese cohort had more TP53 mutation than TCGA cohort. The mutation frequency of TP53 varies from study to study but is usually over 50%. Sinn et al.reported TP53 mutations in 60% of the 368 PDAC patients enrolled in Germany and Austria which is comparable to the frequency of TCGA cohort we reported here (67%) [50]. Lin Shui et al.reported more TP53 mutations in a Chinese PDAC cohort compared to TCGA (62.05% vs 51%) [51]. To be noted, these two studies didn’t specify the subtype of PDAC (head vs body/tail). Another study based on the Chinese population identified 81.8% of patients (n = 154) carrying TP53 mutations [52]. Notably, no significant difference was observed in TP53 mutation frequency between the 85 head and the 69 body/tail PDAC patients in their study. Thus, the ethnic difference could be a potential explanation of different TP53 mutation frequencies. The cohort sizes of the above-mentioned Chinese studies were similar to ours which might lead to cohort bias, further larger sample study is needed to validate the results.
Previous studies suggested BRAF V600E mutations and in-frame deletions near the αC-helix region of the kinase domain are mutually exclusive with KRAS mutations [13, 53]. However, about 0.3–0.4% of KRAS mutant PDACs have concurrent BRAF mutations [32, 54], which explained the concurrent BRAF and KRAS mutations in Chinese cohort. In KRAS wild-type PDAC patients, two novel potentially pathogenic fusions, CHD2 –BRAF and KANK1-MET, were discovered. The former retained an intact BRAF kinase domain, which might cause the activation of BRAF signaling. The intact MET kinase domain of KANK1-MET fusion was fused with the coiled-coils of KANK1, which were located in the N-terminus of KANK1 and reported to be required for KANK1 associated fusion induced cell growth and signaling [55]. It’s worth investigating the efficacy of second-generation BRAF inhibitors and MEK inhibitors in these patients in the future [56].
This study reported, to our knowledge, for the first time the relationship between common tumor biomarker and gene alterations. Further larger sample analysis needs to be performed to verify the results and adjust for possible involvement of other characteristics. Higher preoperative CA125 and CA19-9 were associated with SMAD4 alterations, and elevated CEA was associated with CDKN2A wild-type. Elevated CA125 and CA19-9 were associated with a worse prognosis in PDAC [57, 58]. SMAD4 was also found to be survival-related in several studies and this study [15, 59], whether there are common pathways involved in SMAD4 mutation and elevated tumor biomarkers needs future experiments to clarify. Previous studies suggested CA19-9 levels were correlated to TNM staging as high pre-operation CA19-9 was associated with adverse pathologic features and advanced stage [18], however, no correlation was found in the Chinese cohort. Study has shown as CA19-9 increased, the tumor trends toward unresectable [60]. Since all samples from the cohort were obtained from resected tumors, it is our hypothesize that many high CA19-9 patients were excluded due to the tumor being unresectable, resulting in the irrelevancy. Because of this, the correlation between SMAD4 and CA19-9 was less likely due to the bias of increased tumor staging.
SMAD4 alteration was identified as a predictive marker of short OS in this study. However, the association of SMAD4 and onset age was controversial in previous studies. The study showed that SMAD4 alterations were more enriched in older patients which is consistent with a large-size (n = 3,594) study which suggested patients older than 50 were more likely to harbor SMAD4 alterations [32]. However, Ben-Aharon et al. reported a higher mutational rate of SMAD4 in younger patients with a cutoff of 55 years old [61]. Checking the patients' characteristics between the studies, we found the majority of early-onset patients (80%) in Ben-Aharon et al.’s cohort were of stage IV, which may affect SMAD4 mutation prevalence as loss of SMAD4 can lead to tumor metastases [62, 63].
The two gene alterations, SMAD4 and TGFBR2, included in the established nomogram are partners in the TGF-β signaling pathway [64]. A recent study suggested the predominant function of SMAD4 in collective invasion in PDAC organoids and somatic mutation of TGFBR2 also showed a similar invasion phenotype [65]. Therefore, it’s worth paying attention to the TGF-β signaling pathway in PDAC and investigating the specific mechanism of tumor progression.
Some limitations should be noted. First, the regional effect on PDAC genomic landscape was not taken into account. The epidemiology study showed the prevalence of pancreatic cancer is higher in the East China region [2], but because all patients were selected in a single-center, patients were limited to a certain geographical breadth across China. Secondly, the cohort size is relatively small especially in the situation of lacking published Chinese PDAC genetic information as a reference. Thirdly, the molecular profiling were performed using a targeted panel NGS which is less comprehensive than the WES results of the TCGA database. Finally, due to the study being retrospective, patients lacked the treatment response information.
Conclusions
The present study investigated the genetic landscape of Chinese head PDAC and compared it to the Western population. The study also provided new insights into clinicopathological features and gene alterations. A nomogram was established to predict PDAC prognosis based on tumor genetic alterations and clinical features whose performance was promising in both Chinese and Western cohorts. This study may shed light on the Chinese PDAC molecular profiling and provide a new method to predict prognosis in clinical practice.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- CNV:
-
Copy number variation
- DDR:
-
DNA damage repair
- d-MMR:
-
Mismatch repair deficiency
- FFPE:
-
Formalin-fixed, paraffin-embedded
- MSI:
-
Microsatellite instability
- NGS:
-
Next-generation sequencing
- PDAC:
-
Pancreatic ductal adenocarcinoma
- SNV:
-
Single nucleotide variant
- SV:
-
Structural variants
- TMB:
-
Tumor mutational burden
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32.
Haeberle L, Esposito I. Pathology of pancreatic cancer. Transl Gastroenterol Hepatol. 2019;4:50.
Slack JM. Developmental biology of the pancreas. Development. 1995;121:1569–80.
Artinyan A, Soriano PA, Prendergast C, Low T, Ellenhorn JD, Kim J. The anatomic location of pancreatic cancer is a prognostic factor for survival. HPB (Oxford). 2008;10:371–6.
Lau MK, Davila JA, Shaib YH. Incidence and survival of pancreatic head and body and tail cancers: a population-based study in the United States. Pancreas. 2010;39:458–62.
Lee M, Kwon W, Kim H, Byun Y, Han Y, Kang JS, Choi YJ, Jang JY. The role of location of tumor in the prognosis of the pancreatic cancer. Cancers (Basel). 2020;12:2036.
Winer LK, Dhar VK, Wima K, Morris MC, Lee TC, Shah SA, Ahmad SA, Patel SH. The impact of tumor location on resection and survival for pancreatic ductal adenocarcinoma. J Surg Res. 2019;239:60–6.
Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, Miller DK, Christ AN, Bruxner TJC, Quinn MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52.
Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–3.
Birnbaum DJ, Bertucci F, Finetti P, Birnbaum D, Mamessier E. Head and Body/Tail Pancreatic Carcinomas Are Not the Same Tumors. Cancers (Basel). 2019;11:497.
Dreyer SB, Jamieson NB, Upstill-Goddard R, Bailey PJ, McKay CJ, Biankin AV, Chang DK. Defining the molecular pathology of pancreatic body and tail adenocarcinoma. Br J Surg. 2018;105:e183–91.
Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501.
Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Eshleman JR, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15:4674–9.
Ogura T, Yamao K, Hara K, Mizuno N, Hijioka S, Imaoka H, Sawaki A, Niwa Y, Tajika M, Kondo S, et al. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol. 2013;48:640–6.
Ahmad-Nielsen SA, Bruun Nielsen MF, Mortensen MB, Detlefsen S. Frequency of mismatch repair deficiency in pancreatic ductal adenocarcinoma. Pathol Res Pract. 2020;216:152985.
Luo G, Jin K, Deng S, Cheng H, Fan Z, Gong Y, Qian Y, Huang Q, Ni Q, Liu C, et al. Roles of CA19–9 in pancreatic cancer: Biomarker, predictor and promoter. Biochim Biophys Acta Rev Cancer. 2021;1875:188409.
Meng Q, Shi S, Liang C, Xiang J, Liang D, Zhang B, Qin Y, Ji S, Xu W, Xu J, et al. Diagnostic accuracy of a CA125-based biomarker panel in patients with pancreatic cancer: a systematic review and meta-analysis. J Cancer. 2017;8:3615–22.
Xu HX, Liu L, Xiang JF, Wang WQ, Qi ZH, Wu CT, Liu C, Long J, Xu J, Ni QX, et al. Postoperative serum CEA and CA125 levels are supplementary to perioperative CA19-9 levels in predicting operative outcomes of pancreatic ductal adenocarcinoma. Surgery. 2017;161:373–84.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Lu J, Yu R, Liu R, Liang X, Sun J, Zhang H, Wu H, Zhang Z, Shao YW, Guo J, et al. Genetic aberrations in Chinese pancreatic cancer patients and their association with anatomic location and disease outcomes. Cancer Med. 2021;10:933–43.
Wang H, Ou Q, Li D, Qin T, Bao H, Hou X, Wang K, Wang F, Deng Q, Liang J, et al. Genes associated with increased brain metastasis risk in non-small cell lung cancer: comprehensive genomic profiling of 61 resected brain metastases versus primary non-small cell lung cancer (Guangdong Association Study of Thoracic Oncology 1036). Cancer. 2019;125:3535–44.
Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
Zhang X, Liu F, Bao H, Wang A, Han M, Wu X, Gu Y, Zheng L. Distinct genomic profile in h. pylori-associated gastric cancer. Cancer Med. 2021;10(7):2461.
Xia H, Xue X, Ding H, Ou Q, Wu X, Nagasaka M, Shao YW, Hu X, Ou SI. Evidence of NTRK1 fusion as resistance mechanism to EGFR TKI in EGFR+ NSCLC: results from a large-scale survey of NTRK1 fusions in Chinese patients with lung cancer. Clin Lung Cancer. 2020;21:247–54.
Newman AM, Bratman SV, Stehr H, Lee LJ, Liu CL, Diehn M, Alizadeh AA. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics. 2014;30:3390–3.
R-Core-Team. R: A language and environment for statistical computing. https://www.R-project.org/. Accessed 28 Nov 2021.
Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10:7252–9.
Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, Klempner SJ, Hendifar A, Milind JM, Golan T, et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology. 2019;156:2242–53.
Humphris JL, Patch AM, Nones K, Bailey PJ, Johns AL, McKay S, Chang DK, Miller DK, Pajic M, Kassahn KS, et al. Hypermutation in pancreatic cancer. Gastroenterology. 2017;152:68-74.e62.
Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, Lowery MA, Diaz LA Jr, Mandelker D, Yu KH, et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: challenges and recommendations. Clin Cancer Res. 2018;24:1326–36.
Lupinacci RM, Bachet JB, Andre T, Duval A, Svrcek M. Pancreatic ductal adenocarcinoma harboring microsatellite instability / DNA mismatch repair deficiency. Towards personalized medicine. Surg Oncol. 2019;28:121–7.
Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, Lowery MA, Diaz LA, Mandelker D, Yu KH, et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: challenges and recommendations. Clin Cancer Res. 2018;24:1326–36.
Huang B, Trujillo MA, Fujikura K, Qiu M, Chen F, Felsenstein M, Zhou C, Skaro M, Gauthier C, Macgregor-Das A, et al. Molecular characterization of organoids derived from pancreatic intraductal papillary mucinous neoplasms. J Pathol. 2020;252:252–62.
Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66.
Seidel G, Zahurak M, Iacobuzio-Donahue C, Sohn TA, Adsay NV, Yeo CJ, Lillemoe KD, Cameron JL, Hruban RH, Wilentz RE. Almost all infiltrating colloid carcinomas of the pancreas and periampullary region arise from in situ papillary neoplasms: a study of 39 cases. Am J Surg Pathol. 2002;26:56–63.
Felsenstein M, Noë M, Masica DL, Hosoda W, Chianchiano P, Fischer CG, Lionheart G, Brosens LAA, Pea A, Yu J, et al. IPMNs with co-occurring invasive cancers: neighbours but not always relatives. Gut. 2018;67:1652–62.
Xing C, Zhou W, Ding S, Xie H, Zhang W, Yang Z, Wei B, Chen K, Su R, Cheng J, et al. Reversing effect of ring finger protein 43 inhibition on malignant phenotypes of human hepatocellular carcinoma. Mol Cancer Ther. 2013;12:94–103.
Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16:553–65.
Hobbs GA, Baker NM. Atypical KRAS(G12R) mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 2020;10:104–23.
Diersch S, Wirth M, Schneeweis C, Jörs S, Geisler F, Siveke JT, Rad R, Schmid RM, Saur D, Rustgi AK, et al. Kras(G12D) induces EGFR-MYC cross signaling in murine primary pancreatic ductal epithelial cells. Oncogene. 2016;35:3880–6.
Fakih M, O’Neil B, Price TJ, Falchook GS, Desai J, Kuo J, Govindan R, Rasmussen E, Morrow PKH, Ngang J, et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J Clin Oncol. 2019;37:3003–3003.
Papadopoulos KP, Ou S-HI, Johnson ML, Christensen J, Velastegui K, Potvin D, Faltaos D, Chao RC. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J Clin Oncol. 2019;37:TPS3161–TPS3161.
Kazi A, Xiang S, Yang H, Chen L, Kennedy P, Ayaz M, Fletcher S, Cummings C, Lawrence HR, Beato F, et al. Dual farnesyl and geranylgeranyl transferase inhibitor thwarts mutant KRAS-driven patient-derived pancreatic tumors. Clin Cancer Res. 2019;25:5984–96.
Liu R, Li X, Liu Q, Zhao Z, Yuan H, Ma T. 1540P comprehensive molecular profiling of pancreatic ductal adenocarcinoma in Chinese population. Ann Oncol. 2020;2020(31):S945.
Wang W, Zhou B, Ding Y, Li G, Zheng X, Huang P, Yan S, Li J. 1541P the genomic features of Chinese pancreatic adenocarcinoma and the implications for therapy. Ann Oncol. 2020;2020(31):S945–6.
Sinn M, Sinn BV. TP53 mutations predict sensitivity to adjuvant gemcitabine in patients with pancreatic ductal adenocarcinoma: next-generation sequencing results from the CONKO-001 Trial. Clin Cancer Res. 2020;26:3732–9.
Shui L, Li X, Peng Y, Tian J, Li S, He D, Li A, Tian B, Li M, Gao H, et al. The germline/somatic DNA damage repair gene mutations modulate the therapeutic response in Chinese patients with advanced pancreatic ductal adenocarcinoma. J Transl Med. 2021;19:301.
Zhang X, Feng S, Wang Q, Huang H, Chen R, Xie Q, Zhang W, Wang A, Zhang S, Wang L, et al. Comparative genomic analysis of head and body/tail of pancreatic ductal adenocarcinoma at early and late stages. J Cell Mol Med. 2021;25:1750–8.
Foster SA, Whalen DM, Özen A, Wongchenko MJ, Yin J, Yen I, Schaefer G, Mayfield JD, Chmielecki J, Stephens PJ, et al. Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell. 2016;29:477–93.
Guan M, Bender RJ, Pishvaian MJ, Halverson DC, Tuli R, Klempner SJ, Wainberg ZA, Singhi AD, Petricoin E, Hendifar AE. Molecular and clinical characterization of BRAF mutations in pancreatic ductal adenocarcinomas (PDACs). J Clin Oncol. 2018;36:214–214.
Medves S, Noël LA, Montano-Almendras CP, Albu RI, Schoemans H, Constantinescu SN, Demoulin JB. Multiple oligomerization domains of KANK1-PDGFRβ are required for JAK2-independent hematopoietic cell proliferation and signaling via STAT5 and ERK. Haematologica. 2011;96:1406–14.
Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14:735–48.
Asaoka T, Miyamoto A, Maeda S, Tsujie M, Hama N, Yamamoto K, Miyake M, Haraguchi N, Nishikawa K, Hirao M, et al. Prognostic impact of preoperative NLR and CA19-9 in pancreatic cancer. Pancreatology. 2016;16:434–40.
Liu L, Xu H, Wang W, Wu C, Chen Y, Yang J, Cen P, Xu J, Liu C, Long J, et al. A preoperative serum signature of CEA+/CA125+/CA19-9 >/= 1000 U/mL indicates poor outcome to pancreatectomy for pancreatic cancer. Int J Cancer. 2015;136:2216–27.
Kadera BE, Sunjaya DB, Isacoff WH, Li L, Hines OJ, Tomlinson JS, Dawson DW, Rochefort MM, Donald GW, Clerkin BM, et al. Locally advanced pancreatic cancer: association between prolonged preoperative treatment and lymph-node negativity and overall survival. JAMA Surg. 2014;149:145–53.
Hartwig W, Strobel O, Hinz U, Fritz S, Hackert T, Roth C, Büchler MW, Werner J. CA19-9 in potentially resectable pancreatic cancer: perspective to adjust surgical and perioperative therapy. Ann Surg Oncol. 2013;20:2188–96.
Ben-Aharon I, Elkabets M, Pelossof R, Yu KH, Iacubuzio-Donahue CA, Leach SD, Lowery MA, Goodman KA, O’Reilly EM. Genomic landscape of pancreatic adenocarcinoma in younger versus older patients: does age matter? Clin Cancer Res. 2019;25:2185–93.
Wang Y, Chu J, Yi P, Dong W, Saultz J, Wang Y, Wang H, Scoville S, Zhang J, Wu LC, et al. SMAD4 promotes TGF-β-independent NK cell homeostasis and maturation and antitumor immunity. J Clin Invest. 2018;128:5123–36.
Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–73.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84.
Huang W, Navarro-Serer B, Jeong YJ. Pattern of invasion in human pancreatic cancer organoids is associated with loss of SMAD4 and clinical outcome. Cancer Res. 2020;80:2804–17.
Acknowledgements
The authors thank Technicians Lu Jiang and Xue Han for technical support.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 81773109], the Natural Science Foundation of Jiangsu Province [grant number BK20151582], National key Clinical Specialty Construction Project (2014), Joint key project funded by Southeast University and Nanjing Medical University [grant numbers 2242019K3DN09, 2019DN0011] and the Fund of the priority Academic Programme Development of Jiangsu Higher Education Institution [grant number JX1023-1801].
Author information
Authors and Affiliations
Contributions
Y.Y. and Y.D. designed the study and drafted the paper. Y.Y., Y.D., Y.G., S.Z., M.L., X.L., G.S., B.Z. were involved in specimens and information collection. J.P. and G.S. performed most of the experiments. Y.Y., Y.D., L.Z., Y.S. were involved in the sequencing data processing. L.Z. and Y.S. analyzed the TCGA database. Y.Y. and J.L were involved in statistical analysis. Y.M., Q.O., X.W. were involved in modifications and corrections of the manuscript. Z.Z. supervised and acquired funds for the research. All authors reviewed the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University(2020-SR-273). Written consent was obtained from all participants. The study was performed in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
List of 425 targeted sequencing genes.
Additional file 2: Table S2.
List of 91 TCGA patients' barcode.
Additional file 3: Table S3.
List of all detected alterations and TMB in Chinese cohort.
Additional file 4: Figure S1.
Survival curve of KRAS G12 subtypes, MS status, and TMB subgroups.
Additional file 5: Figure S2.
The association between cancer antigen levels and gene alterations.
Additional file 6: Figure S3.
Vascular invasion possibility is associated with RNF43.
Additional file 7: Figure S4.
Gene rearrangements detected in two KRAS wild-type patients.
Additional file 8: Table S4.
Clinical and pathological data of Chinese cohort.
Additional file 9: Table S5.
FFPE DNA concentrations of Chinese cohort.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, Y., Ding, Y., Gong, Y. et al. The genetic landscape of pancreatic head ductal adenocarcinoma in China and prognosis stratification. BMC Cancer 22, 186 (2022). https://doi.org/10.1186/s12885-022-09279-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-09279-9