
Abstract
Background
Neuroblastoma (NB) patients with MYCN amplification or overexpression respond poorly to current therapies and exhibit extremely poor clinical outcomes. PI3K-mTOR signaling-driven deregulation of protein synthesis is very common in NB and various other cancers that promote MYCN stabilization. In addition, both the MYCN and mTOR signaling axes can directly regulate a common translation pathway that leads to increased protein synthesis and cell proliferation. However, a strategy of concurrently targeting MYCN and mTOR signaling in NB remains unexplored. This study aimed to investigate the therapeutic potential of targeting dysregulated protein synthesis pathways by inhibiting the MYCN and mTOR pathways together in NB.
Methods
Using small molecule/pharmacologic approaches, we evaluated the effects of combined inhibition of MYCN transcription and mTOR signaling on NB cell growth/survival and associated molecular mechanism(s) in NB cell lines. We used two well-established BET (bromodomain extra-terminal) protein inhibitors (JQ1, OTX-015), and a clinically relevant mTOR inhibitor, temsirolimus, to target MYCN transcription and mTOR signaling, respectively. The single agent and combined efficacies of these inhibitors on NB cell growth, apoptosis, cell cycle and neurospheres were assessed using MTT, Annexin-V, propidium-iodide staining and sphere assays, respectively. Effects of inhibitors on global protein synthesis were quantified using a fluorescence-based (FamAzide)-based protein synthesis assay. Further, we investigated the specificities of these inhibitors in targeting the associated pathways/molecules using western blot analyses.
Results
Co-treatment of JQ1 or OTX-015 with temsirolimus synergistically suppressed NB cell growth/survival by inducing G1 cell cycle arrest and apoptosis with greatest efficacy in MYCN-amplified NB cells. Mechanistically, the co-treatment of JQ1 or OTX-015 with temsirolimus significantly downregulated the expression levels of phosphorylated 4EBP1/p70-S6K/eIF4E (mTOR components) and BRD4 (BET protein)/MYCN proteins. Further, this combination significantly inhibited global protein synthesis, compared to single agents. Our findings also demonstrated that both JQ1 and temsirolimus chemosensitized NB cells when tested in combination with cisplatin chemotherapy.
Conclusions
Together, our findings demonstrate synergistic efficacy of JQ1 or OTX-015 and temsirolimus against MYCN-driven NB, by dual-inhibition of MYCN (targeting transcription) and mTOR (targeting translation). Additional preclinical evaluation is warranted to determine the clinical utility of targeted therapy for high-risk NB patients.
Similar content being viewed by others


Background
Neuroblastoma (NB) is the most common extracranial pediatric solid tumor of neural crest origin and accounts for approximately 10% of childhood cancers and 15% of cancer-related deaths in children. Approximately 50% of NB patients are diagnosed with high-risk disease, and despite intensive multimodal therapy options (including radiation, surgery, and chemotherapy), effective treatment for these patients remains elusive [1, 2]. Particularly, amplification of the neural MYC (MYCN) oncogene, which occurs in 20–30% of all NB tumors and nearly 50% of the high- risk cases, remains a key predictor of poor outcomes. MYCN-amplified NB tumors typically exhibit high malignancy, metastatic properties, and treatment resistance [3, 4]. Therefore, upstream and downstream regulatory components of the MYCN-driven tumorigenic programs contain promising targets for the identification of novel therapeutics for these high-risk patients.
One of the most frequently deregulated oncogenic pathways in cancers, is the protein synthesis (translation) pathway that drives increased cell proliferation and cancer progression/resistance [5, 6]. Similar to MYC protein, MYCN plays an important role in protein synthesis by controlling the transcription of several components of protein synthesis machinery including components involved in mRNA translation and ribosome biogenesis [7,8,9,10]. Similar to MYC protein, MYCN itself is considered to be an undruggable target because of its short half-life and complex protein structure; however, targeting epigenetic regulators of MYCN provides a promising alternative strategy [11, 12]. Bromodomain and extra-terminal (BET) family proteins have been shown to promote MYCN transcription. In preclinical studies, inhibiting BET protein function has shown promise as a therapeutic strategy to target MYCN in NB and other cancers [13,14,15,16,17].
mTOR signaling is another key regulator of protein synthesis, which is frequently deregulated in cancers including NB [18,19,20]. MTOR kinase regulates protein synthesis by phosphorylating key translation factors (4EBP1/eIF4E) upstream of the translation initiation complex [18]. Notably, it has been shown that mTOR signaling can stabilize MYCN protein levels by inducing MYCN translation [21]. Together, these observations suggest the potential to block deregulated MYCN-driven proliferation by co-delivering drugs that target global transcription and translation.
We hypothesize that combined inhibition of transcription (by BET-protein inhibition) and translation (by mTOR inhibition) will synergistically blockade global protein synthesis and proliferation in MYC-driven NB tumor cells. Using small molecule/pharmacologic approaches, we tested this hypothesis by targeting BET with JQ1 or OTX-015 and mTOR with temsirolimus, in NB cell lines.
Methods
Cell lines and inhibitors
Non-MYCN-amplified NB cell lines (SK-K-AS, SK-N-SH) and MYCN-amplified NB cell lines (SK-N-BE2, IMR-32, and SK-N-DZ) were purchased from American Type Culture Collection (USA). Non-MYCN-amplified NB cell line CHLA-255 was provided by Dr. Kishore Challagundla (UNMC). The identity of cell lines was confirmed by their respective cell bank using STR analyses. Cell lines were also verified for mycoplasma-free condition using the MycoSensor-PCR assay kit (Agilent-Technologies, USA). Cell lines were cultured in Eagle’s Minimal Essential Medium (EMEM) or Roswell Park Memorial Institute (RPMI)-1640 media containing 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen Life Technologies, USA). Experiments were performed under 8–10 passages for each cell line. Small molecule inhibitors (JQ1, OTX-015 and temsirolimus) and cisplatin (a chemotherapeutic drug) were purchased from Sellekchem LLC (USA).
Cell viability assay
Effects of inhibitors on NB Cell growth/viability were assessed using the MTT assay as previously described [22, 23].
Neurosphere assay
Effects of inhibitors on NB spheres were performed using the neurosphere/sphere assay as previously described [22].
Cell cycle distribution and apoptosis analyses
Analysis of cell cycle distribution in NB cells was performed using a propidium iodide (PI staining) flow cytometry kit (Abcam, UK) according to manufacturer’s instructions. Apoptosis in NB cells was assessed using an Annexin-V flow-cytometry assay kit (BD-Biosciences, USA) following the manufacturer’s instructions. The flow cytometry analysis of Annexin-V/PI stained cells was determined using the FACSCalibur cell sorter system (BD Biosciences, USA).
Global protein synthesis assay
NB cells were treated with inhibitors alone or in combination in 96-well plates (2 × 104 cells/well) for 24 h. After treatment, culture media was replaced with fresh media containing O-propargyl-puromycin (OPP) and incubated for 30 min at 37 °C to be incorporated in translating polypeptide chains. Cells were then fluorescently stained with 5-FAM-Azide. The detection of fluorescent-labelled OPP was performed using the Protein Synthesis Assay Kit (#601100, Cayman Chemical, USA), according to the manufacturer’s instructions.
Immunoblotting
Western blot analysis of the inhibitor-treated NB cells was performed as described previously [23]. Primary and secondary antibodies used in this analysis included MYCN (Cell Signaling Technology #9405), BRD4 (Cell Signaling Technology #13440), p-4EBP1 (Ser65, Cell Signaling Technology #9456) 4EBP1 (Cell Signaling Technology #9452), p-eIF4E (Ser209, Cell Signaling Technology #9741), eIF4E (Cell Signaling Technology #9742), p-S6K (Thr421/Ser424, Cell Signaling Technology #9204), S6K (Cell Signaling Technology #9202), Nestin (Santacruz Biotechnology #sc-23,927), SOX2 (Cell Signaling Technology #3579), GAPDH (Cell Signaling Technology #2118), Cyclophillin B (Cell Signaling Technology #43603), CD133 (BD Biosciences) and HRP-conjugated secondary antibodies (anti-Rabbit/Mouse, Jackson ImmunoResearch Laboratory).
Statistical analysis
Each experiment was repeated at last an additional three times and the mean ± standard error values calculated. Statistical significance (p-value) was analyzed using two-tailed Student’s t-test or analysis of variance (ANOVA) and p-values > 0.05 considered significant. GraphPad Prism-V6 software was used to determine IC50 values and dose-response curves of inhibitors in NB cell lines. The Chou-Talalay combination index (CI) method was used to analyze synergy/interaction between inhibitors by using CalcuSyn software (Biosoft, UK). CI < 0.9 indicates synergism, 0.9–1.1 additivity and > 1.1 antagonism.
Results
Synergistic effects of JQ1 and temsirolimus on NB cell growth
We used small molecule inhibitors JQ1 and temsirolimus (TEM, hereafter) to target MYCN transcription (BET proteins) and mTOR signaling, respectively, [24, 25] in NB. Using the MTT assay, we first determined the IC50 of each inhibitor on cell viability of three non-MYCN- and three MYCN-amplified NB cell lines. Our results showed that as single agents, JQ1 and TEM inhibited NB cell growth with relatively lower IC50 values in MYCN-amplified NB cell lines (Table 1), suggesting superior efficacy of each inhibitor against MYCN-driven NB cell lines, compared to non-MYCN-amplified NB cell lines.
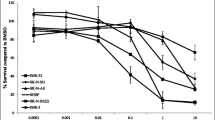
We next tested the efficacy of combined JQ1/TEM to explore potential for synergistic growth inhibition on NB cells. NB cell lines, including three MYCN-amplified and two non-MYCN-amplified NB cell lines, were treated with increasing concentrations of JQ1 and TEM alone or in combination for 72 h. As shown in Fig. 1, co-treatment of JQ1/TEM significantly suppressed growth of all NB cell in a dose-dependent manner, compared with single agent treatment. Again, this co-treatment had greater on growth inhibition effects on MYCN-amplified cell lines, compared to non-MYCN-amplified cell line. The combination index (CI) analyses by Chou-Talalay method [26] confirmed that combination of JQ1/TEM had strong synergistic inhibitory effects on NB cell growth, with CI values ranging 0.3–0.8. These results suggested synergistic anti-NB potential of MYCN/mTOR inhibition.

Synergistic effects of JQ1 and temsirolimus (TEM) on NB cell growth. Cell viability (MTT) assay showing dose-dependent growth effects of JQ1/TEM in NB cell lines at 72 h. Viable cells (%) is relative to DMSO-treated cells. Values represent mean ± SEM. Bar graphs show combination index (CI) analyses for the synergism of JQ1 and TEM in NB cell lines
Co-treatment with JQ1 and TEM induces cell cycle arrest and apoptosis
To determine combined effects of JQ1 and TEM on cell cycle and apoptosis, two highly MYCN-amplified NB (SK-N-BE2, SK-N-DZ) cell lines were treated with a suboptimal concentration of each inhibitor alone or in combination and subjected to cell cycle and apoptosis analyses using propidium-iodide and Annexin-V staining, respectively, followed by flow cytometry. The cell cycle analyses in both MYCN-amplified cell lines revealed that JQ1 and TEM alone slightly caused cell cycle arrest in G1 phase, while co-treatment with JQ1 and TEM drastically arrested the cells in G1 phase (Fig. 2a). The Annexin-V assay demonstrated that treatment with JQ1 or TEM alone increased the percentage of apoptotic cells. However, the co-treatment with JQ1 and TEM resulted in significant further induction of apoptosis in both NB lines (Fig. 2b) and showed consistency with the results of the MTT growth study. These results suggest that the combination of these two inhibitors suppresses growth and/or survival of MYCN-amplified NB cells in vitro.

Combined effects of JQ1 and TEM on cell cycle arrest and apoptosis. (a) Representative flow cytometry plots show cell cycle distribution in SK-N-BE2 cells treated with JQ1 (0.5 μM) and TEM (2 μM) alone or combined for 24 h. On the right, graphs show the quantification of cell cycle distribution in two MYCN-amplified (SK-N-BE2, SK-N-DZ) NB cell lines treated with JQ1 (0.5 μM) and TEM (2 μM) alone or combined for 24 h. (b) Representative flow scatter diagrams show apoptosis induction in SK-N-BE2 cells treated with JQ1 (0.5 μM) and TEM (2 μM) alone or combined for 72 h. On the right, bar graphs show the quantification of Annexin-V/PI double positive apoptotic cells in two MYCN-amplified (SK-N-BE2, SK-N-DZ) and one non-MYCN-amplified (SK-N-AS) NB cell lines treated with JQ1 (0.5 μM) and TEM (2 μM) alone or combined for 72 h. Values, mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (Student-t-test)
Co-treatment with JQ1 and TEM downregulates the expression of MYCN and mTOR signaling components
To establish the molecular mechanism(s) associated with JQ1/TEM anti-NB activity, we examined the expression/activation of key components of MYCN and mTOR signaling pathways by western blotting in SK-N-BE2 and SK-N-DZ cell lines. Single agent treatments of MYCN-amplified cells with JQ1 or TEM significantly suppressed expression of MYCN and BET protein BRD4 and downregulated the levels of phosphorylated/activated signaling proteins (p-S6K, p-4EBP1, and p-eIF4E) of the mTOR (translation) pathway (Fig. 3a and b). Co-treatment with JQ1 and TEM further downregulated the expression of the above mentioned mTOR signaling components, as well as MYCN expression, compared with single agent treatments. These data suggest that concurrent inhibition of MYCN transcription and mTOR signaling cooperatively suppresses the protein synthesis pathway, justifying why this combined inhibition exerts the greatest antitumor effects in MYCN-amplified NB.

Combined effects of JQ1 and TEM on target pathways/molecules and global protein synthesis. (a) Western blot images for the expression of key components of MYCN/mTOR signaling in two MYCN-amplified NB cell lines following treatment with JQ1 (0.5 μM) and TEM (2 μM) alone and combined for 24 h. The original uncropped images of these blots are provided in the Additional File 1 (Fig. S1 and S2). (b) Bar graphs show the quantification of expression of key target proteins (shown in western blot images) relative to the control (DMSO) in the combined blots of SK-N-BE2 and SK-N-DZ cells after GAPDH (loading control) normalization using Image J software. The values represent the mean ± SEM of three replicates of blot. *p < 0.05; **p < 0.01; ***p < 0.001 (Student t test, vehicle/ or single agents vs. combination). (c) Overall protein synthesis measurement by OPP-incorporation following treatment with JQ1 (0.5 μM) and TEM (2 μM) alone or in combination for 24 h. CHX (50 μg/ml, 1 h) was used as a positive control for protein synthesis inhibition. Values represent mean ± SEM. **p < 0.01 (Student-t-test)
Co-treatment with JQ1 and TEM inhibits global protein synthesis
Because of the key role of MYCN/mTOR signaling in controlling global protein synthesis, we further tested whether the inhibition of MYCN/mTOR represented a global blockage of protein synthesis. To this end, we performed protein synthesis assay using a robust chemical method based on a cell permeable analogue of puromycin, O-Propargyl-puromycin (OPP), in SK-N-BE2 and SK-N-DZ cell lines treated with JQ1 and TEM alone or in combination. Incorporation of OPP to nascent polypeptide chains can be labeled via copper catalyzed click-chemistry using 5-FAMAzide in order to quantify total protein synthesis. In this assay, we used cycloheximide as the positive control for protein synthesis inhibition. This fluorescence-based assay displayed high protein synthesis in control solvent (DMSO)-treated cells and strong inhibition of protein synthesis when blocked with cycloheximide (Fig. 3c). Although treatments of JQ1 and TEM alone showed strong inhibitory effects on protein synthesis, the combination of these two caused significant further reduction in total protein synthesis, resulting in the lowest fluorescent signal in both NB cell lines and suggesting a synergistic effect of the MYCN and mTOR targeting on global protein synthesis.
Combined effect of JQ1 and TEM on neurosphere formation
We next investigated the effect of JQ1 and TEM, alone or combined, in a neurosphere model of MYCN-amplified NB cells. Figure 4a shows micrographs of sphere formation in SK-N-BE2 cells. Treatment with JQ1 or TEM alone significantly inhibited sphere formation, with further reduction of sphere formation when both were combined (Fig. 4b). We further tested the effects of JQ1 and TEM on the expression of neural stem cell markers (Nestin, CD133, SOX2) in SK-N-BE2 spheres by western blot analysis. We observed that JQ1 and TEM, either alone or combined, strongly inhibited the expression of these stem cell markers and inhibited MYCN expression (Fig. 4c). These data suggest that combined inhibition of MYC transcription and mTOR signaling has anti-tumor effects on neurospheres and associated stem cell markers.

Combined effects of JQ1 and TEM on neurosphere formation. (a) Representative micrograph of spheres of SK-N-BE2 cells with the treatment of control-(DMSO) or JQ1 (0.5 μM) and TEM (2 μM) alone or combined for 7 days. Scale bar, 1000 μm. (b) Quantification of the number of neurospheres following treatments. The values represent mean ± SEM. **p < 0.01; ***p < 0.001 (Student-t-test). (c) Western blot analysis for the expression of neural stem cell markers following treatment of neurospheres with JQ1 (0.5 μM) and TEM (2 μM) alone or combined for three days. Cyclophilin was used as the loading control in this analysis. The original uncropped images of these blots are provided in the Additional File 1 (Fig. S3)
JQ1 and TEM chemosensitizes NB cells
Given the limited success of current therapies, we next sought to determine whether JQ1 or TEM could enhance the anti-NB efficacy of chemotherapy by sensitizing NB cells. Cisplatin is one of the most common chemotherapeutic drugs in the treatment of NB patients. To evaluate the enhanced efficacy of inhibitors on cisplatin-mediated NB cytotoxicity, we treated NB (SK-N-AS, SK-N-BE2, SK-N-DZ) cell lines with cisplatin and either JQ1, TEM, or the combination cisplatin alone or in combination, in stepwise doses. At 72 h we determined cell growth using the MTT assay. Results shown in Fig. 5 clearly show that co-treatment of NB cell lines with inhibitors (JQ1 or TEM) and cisplatin significantly inhibited cell growth in a dose-dependent manner, compared to cisplatin and inhibitors alone. Combination index analyses further show that JQ1 or TEM synergistically increased the cytotoxicity of cisplatin in all NB cell lines tested. Of these combinations, JQ1 demonstrated a significantly greater efficacy in enhancing cisplatin-mediated NB cytotoxicity. Results also indicated a higher sensitivity of MYCN-amplified NB cells to these combined treatments, compared to non-MYCN-amplified SK-N-AS cells. In summary, these data show that JQ1 or TEM either combined together or individually combined with cisplatin chemotherapy, synergistically inhibits NB cell growth.

JQ1 and TEM chemosensitize NB cells. MTT results showing the dose-dependent effects of JQ1 and TEM alone or combined with cisplatin (Cis) chemotherapy in NB cell lines at 72 h, as indicated. The values represent the means ± SEM. Bar graphs show combination index (CI) analyses for the synergism between JQ1/Cis or TEM/Cis in NB cell lines
Synergistic anti-cancer efficacies of OTX-015 and TEM in NB cells
In previous experiments, the rationale for using JQ1 was its advantages over other BET inhibitors in preclinical cancer studies. Preclinical studies with JQ1 offered a great opportunity to better understand the biology of BET proteins and validate these proteins as the anti-cancer targets [14, 24]. In addition, JQ1 has been shown to efficiently target MYC/MYCN transcription in pediatric NB and medulloblastoma [15,16,17]. However, JQ1 is not being considered in clinical trials because of its short half-life [24]. As the proof of concept that if the combined inhibition of BET protein and mTOR signaling can be translated into the clinic, we also utilized a clinically relevant BET protein inhibitor OTX-015 (OTX, hereafter) which is currently in clinical trials for several advanced cancers [14]. Using previously reported doses of OTX in NB cell lines [16], we tested its combination efficacy with TEM in two MYCN-amplified NB cell lines. As shown in Fig. 6a, co-treatment of OTX and TEM significantly suppressed growth of NB cell lines in a dose-dependent manner, compared to single agents alone. CI analyses further confirmed that combination of OTX and TEM had a highly synergistic inhibitory effect on NB cell growth with a CI value below 0.7. The results from cell cycle (PI staining) and apoptosis (Annexin-V/PI staining) analyses in NB cells showed that the combination significantly induced G1 cell cycle arrest and apoptosis (Fig. 6b and c), compared to single agents. Our western blot results showed that compared to single agent treatments, combination of OTX and TEM significantly suppressed expression of MYCN and downregulated the levels of key downstream targets (phosphorylated-4EBP1/eIF4E) of the mTOR pathway (Fig. 6d). The results from global protein synthesis investigation using OPP-based assay, further demonstrated synergistic inhibition of general protein synthesis rate by OTX and TEM, compared to single agents (Fig. 6e). These results consistently suggested the importance of co-targeting BET protein and mTOR signaling in NB.

Synergistic effects of OTX-015 (OTX) and TEM on NB cell growth. (a) Cell viability (MTT) assay showing dose-dependent growth effects of OTX/TEM in two MYCN-amplified NB cell lines at 72 h. Viable cells (%) is relative to DMSO-treated cells. Values represent mean ± SEM. Bar graphs show combination index (CI) analyses for the synergism of OTX and TEM in NB cell lines. (b) Analysis of cell cycle distribution in SK-N-BE2 cells treated with OTX (1 μM) and TEM (2 μM) alone or combined for 24 h. (c) Quantification of Annexin-V/PI positive apoptotic cells in two MYCN-amplified (SK-N-BE2, SK-N-DZ) NB cell lines treated with OTX (1 μM) and TEM (2 μM) alone or combined for 72 h. Values, mean ± SEM. **p < 0.01; ***p < 0.001 (Student-t-test). (d) Western blot images for the expression of key components of MYCN/mTOR signaling in two MYCN-amplified NB cell lines following treatment with OTX (1 μM) and TEM (2 μM) alone and combined for 24 h. Cyclophilin was used as the loading control in these analyses. The original uncropped images of these blots are provided in the Additional File 1 (Fig. S4). (e) Global protein synthesis measurement by OPP-incorporation following treatment with OTX (1 μM) and TEM (2 μM) alone or in combination for 24 h. CHX (50 μg/ml, 1 h) was used as a positive control for protein synthesis inhibition. Values represent mean ± SEM. **p < 0.01
Discussion
Despite the availability of intensive multimodal therapy, the prognosis for patients with MYCN-amplified NB remains extremely poor [4]. Since MYCN is not directly druggable, indirectly inhibiting it by targeting upstream or downstream components of the MYCN pathway that can regulate its oncogenic activities might be the ideal option [10, 12]. As a transcription factor, MYCN has tight control over global transcription and translation [9]. Aberrant activation of several oncogenic pathways, including MYC pathway, is a hallmark of many childhood cancers, including NB [10]. MTOR signaling is one of the key oncogenic pathways which is often deregulated in MYC-driven cancers, including NB [20]. Moreover, it has been shown that MYC proteins and the mTOR pathway cooperate with each other at the transcription and translation levels, respectively, to elevate overall protein synthesis rate, leading to increased cell proliferation and cancer progression [21, 22, 27]. These pathways, either working individually or together, can cause cancer progression. However, inhibition of one pathway alone will often result in minor to moderate anti-tumor activity, suggesting concurrent targeting of MYCN transcription and mTOR signaling is important strategy to enhance efficacy. Here, using small molecule therapeutics, we show that combined inhibition of MYCN transcription by BET inhibitors (JQ1/OTX) and mTOR signaling by TEM, synergistically inhibited NB cell growth and survival. The combined activity was more potent than inhibition of either single pathway.
BET protein inhibitors exert biological effects by competitively binding to the acetyl-lysine pockets and displacing BET proteins from chromatin, leading to the repression of oncogene transcription. BET protein inhibitors have been shown to target MYC/MYCN transcription in several cancers, including NB, demonstrating their potential as preclinical anticancer agents [13,14,15,16,17]. In comparison, mTOR inhibitors, another class of anticancer agents, exert their inhibitory effects by blocking the activation of mTOR signaling, leading to inhibited translation of proteins, including MYCN protein [21, 25]. Several inhibitors of BET protein and mTOR signaling either alone or in combination with other target agents, are in multiple clinical trials in patients with advanced cancers [14, 25]. In this study, we used JQ1, a potent preclinical BET protein inhibitor, as a proof of concept for targeting MYCN transcription [16]. Since JQ1 is not being tested in clinical trials due to its short half-life [28], in parallel, we used the clinically relevant BET protein inhibitor OTX-015 as an alternative agent to target MYCN. We confirmed that JQ1 and OTX-015 have similar anti-NB efficacy in our proof-of-concept study. To target mTOR signaling we used the clinically relevant mTOR inhibitor TEM [25], an FDA approved drug for the treatment of renal cell carcinoma. These inhibitors have demonstrated promising antitumor activities against multiple solid and hematological cancers [14, 25]. Here, we identify a synergistic interaction between BET and mTOR inhibitors to induce antitumor effects in NB cells. In addition, our evidence of cell cycle arrest, induction of apoptosis and inhibition of NB neurospheres further reinforce the clinical relevance of our combination approach.
In addition, our data provide insights into the molecular mechanism(s) underlying the antitumor efficacy of JQ1/TEM we observed in NB. We show that co-treatment with JQ1 and TEM synergistically reduce the expression of key component proteins of the activated mTOR translation pathway, along with suppression of MYCN and BRD4 (a key member of BET family proteins) protein expression in NB cells. We further show that this co-inhibition of MYCN/mTOR by JQ1/TEM results in significant blockade of global protein synthesis in NB cells. These observations in NB are consistent with our previous study in medulloblastoma where we have shown similar combined activity of JQ1/TEM on MYC/mTOR targets, leading to inhibition of global protein synthesis. Overall, these findings suggest a synergistic and cooperative activity of JQ1/TEM on MYCN and mTOR pathways in aggressive NB.
While our data show that although individual or concomitant inhibition of BET/mTOR has greater antitumor efficacies against MYCN-amplified NB cells, the combination also efficiently inhibits cell growth and induces apoptosis in non-MYCN-amplified NB cells, pointing to a broader relevance of our combined approach in NB. Because non-MYCN-amplified NB cell lines, including SK-N-AS, express MYC [29] which is a target for combined BET/mTOR in medulloblastoma cells [22]. MYC may be a secondary target for these inhibitors in the context of NB.
In NB and other cancers, MYCN and mTOR signaling have been shown to play key roles in cancer stem cells and contribute to relapse and drug-resistance. NB cells express neural stem cell markers such as CD133 and Nestin and have the ability to form neurospheres [30,31,32,33]. Our initial findings with the anti-NB efficacy of MYCN/mTOR inhibition on neurospheres and stem cell markers indicate that individual or combined inhibition of MYCN/mTOR might target cancer stem cells - potentially minimizing recurrence of NB. Mounting evidence suggest that increased protein synthesis in cells not only correlates with increased cell proliferation/survival, but also often involves in stem cell fate, including neural stem cell markers [34, 35]. In line with these, our data is consistent that cooperative inhibition of MYCN/mTOR by proposed drugs downregulates the expression/activation of MYCN/mTOR key targets and inhibits global protein synthesis, thereby enables cell growth/cell cycle arrest, apoptosis and inhibits neurosphere formation/stem cell markers.
Since overexpression or amplification of MYC/MYCN and activation of mTOR signaling are often associated with chemoresistance in many cancers including NB [36,37,38], we also wanted to see whether inhibition of these signaling pathways helps to chemosensitize NB cells. Our results revealed that both JQ1 and TEM significantly enhanced cisplatin-induced growth inhibition in MB cells, suggesting that inhibition of MYCN/mTOR signaling not only inhibits cell proliferation and survival of NB cells, but also sensitizes NB cells to chemotherapy. The exact mechanism of synergy between these inhibitors and cisplatin requires further investigation. However, activation of both MYC and PI3K-mTOR signaling pathways have been shown to be associated with platinum-based therapy resistance [39,40,41]. The cytotoxicity of cisplatin involves the damaging of DNA replication/repair mechanisms. Similarly, targeting MYC and mTOR signaling have been shown to induce DNA damage response and contribute susceptibility to cisplatin-induced cell death. Therefore, co-induction of DNA damage by cisplatin and JQ1 or TEM could be the potential mechanism of observed synergy.
In clinical trials studies, TEM alone or in combination with other clinical agents, such as chemotherapeutic drugs temzilomide and irrinotican, has shown no significant benefit for relapsed NB patients [42, 43]. This suggests further exploration of combining TEM with other targeted therapies. Our data indicate that one of potential combinatorial target could be the inhibition of BET proteins. Since inhibitors of BET proteins, including OTX and structurally similar inhibitors to JQ1, are currently in clinical trials for several cancers, it is more likely that combination of TEM with BET inhibitors can be translated in clinic for NB and other MYC/MYCN-driven cancers therapies.
Conclusions
Our study demonstrates that targeting dysregulated protein synthesis pathway by pharmacologic dual-inhibition of MYCN transcription (by BET protein inhibition) and mTOR signaling has significant preclinical anti-NB efficacies in inducing cell growth inhibition, cell cycle arrest and apoptosis in vitro. Combination of JQ1 or OTX with TEM synergistically inhibited global protein synthesis by downregulating the key components and downstream targets of MYCN/mTOR signaling. Thus, this study is the first to demonstrate synergy in the combined inhibition of BET protein and mTOR signaling in NB. Our study also revealed that inhibition of MYCN/mTOR not only inhibits cell growth and survival in NB cells, but also chemosensitizes NB cells. Beyond NB, this therapeutic approach can be of broader relevance for therapy of MYC/MYCN-addicted cancers, as we have previously shown a synergistic antitumor efficacy of BET/mTOR inhibitors in MYC-driven medulloblastoma [22]. While further studies using appropriate in vivo models are needed to evaluate this combination, our study highlights a basis for considering this combination approach as a new therapy for NB.
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Abbreviations
- BET:
-
Bromodomain extra-terminal
- CI:
-
Combination index
- FBS:
-
Fetal bovine serum
- h:
-
Hour
- IC50 :
-
Inhibitory concentration of inhibitor with 50% inhibition
- NB:
-
Neuroblastoma
- mTOR:
-
Mammalian Target of Rapamycin
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;
- MYC:
-
v-myc avian mylocytomatosis viral oncogene homolog
- MYCN:
-
v-myc avian mylocytomatosis viral oncogene neuroblastoma homolog
- OPP:
-
O-Propargyl-puromycin
- OTX:
-
OTX-015
- TEM:
-
Temsirolimus
- μM:
-
Micromolar
References
Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362(23):2202–11. https://doi.org/10.1056/NEJMra0804577.
Tolbert VP, Matthay KK. Neuroblastoma: clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018;372(2):195–209. https://doi.org/10.1007/s00441-018-2821-2.
Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers. 2016;2(1):16078. https://doi.org/10.1038/nrdp.2016.78.
Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013;3(10):a014415. https://doi.org/10.1101/cshperspect.a014415.
Truitt ML, Ruggero D. New frontiers in translational control of the cancer genome. Nat Rev Cancer. 2017;17(5):332. https://doi.org/10.1038/nrc.2017.30.
Robichaud N, Sonenberg N, Ruggero D, Schneider RJ. Translational Control in Cancer. Cold Spring Harb Perspect Biol. 2019;11(7):11(7). https://doi.org/10.1101/cshperspect.a032896.
Ruggero D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009;69(23):8839–43. https://doi.org/10.1158/0008-5472.CAN-09-1970.
van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10(4):301–9. https://doi.org/10.1038/nrc2819.
Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001;20(6):1383–93. https://doi.org/10.1093/emboj/20.6.1383.
Liu R, Shi P, Wang Z, Yuan C, Cui H. Molecular mechanisms of MYCN Dysregulation in cancers. Front Oncol. 2021;10:625332. https://doi.org/10.3389/fonc.2020.625332.
Wolpaw AJ, Bayliss R, Büchel G, Dang CV, Eilers M, Gustafson WC, et al. Drugging the “Undruggable” MYCN oncogenic transcription factor: overcoming previous obstacles to impact childhood cancers. Cancer Res. 2021;81(7):1627–32. https://doi.org/10.1158/0008-5472.CAN-20-3108.
Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3(1):5. https://doi.org/10.1038/s41392-018-0008-7.
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17. https://doi.org/10.1016/j.cell.2011.08.017.
Stathis A, Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov. 2018;8(1):24–36. https://doi.org/10.1158/2159-8290.CD-17-0605.
Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20(4):912–25. https://doi.org/10.1158/1078-0432.CCR-13-2281.
Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, et al. Targeting MYCN-driven transcription by BET-Bromodomain inhibition. Clin Cancer Res. 2016;22(10):2470–81. https://doi.org/10.1158/1078-0432.CCR-15-1449.
Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3(3):308–23. https://doi.org/10.1158/2159-8290.CD-12-0418.
Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda). 2006;21:362–9. https://doi.org/10.1152/physiol.00024.2006.
Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. https://doi.org/10.1038/nature10912.
Mei H, Wang Y, Lin Z, Tong Q. The mTOR signaling pathway in pediatric neuroblastoma. Pediatr Hematol Oncol. 2013;30(7):605–15. https://doi.org/10.3109/08880018.2013.798058.
Vaughan L, Clarke PA, Barker K, Chanthery Y, Gustafson CW, Tucker E, et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget. 2016;7(36):57525–44. https://doi.org/10.18632/oncotarget.10544.
Chaturvedi NK, Kling MJ, Griggs CN, Kesherwani V, Shukla M, McIntyre EM, et al. A novel combination approach targeting an enhanced protein synthesis pathway in MYC-driven (group 3) Medulloblastoma. Mol Cancer Ther. 2020;19(6):1351–62. https://doi.org/10.1158/1535-7163.MCT-19-0996.
Chaturvedi NK, Rajule RN, Shukla A, Radhakrishnan P, Todd GL, Natarajan A, et al. Novel treatment for mantle cell lymphoma including therapy-resistant tumor by NF-kB and mTOR dual-targeting approach. Mol Cancer Ther. 2013;12(10):2006–17. https://doi.org/10.1158/1535-7163.MCT-13-0239.
Shorstova T, Foulkes WD, Witcher M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer. 2021;124(9):1478–90. https://doi.org/10.1038/s41416-021-01321-0.
Tian T, Li X, Zhang J. mTOR signaling in Cancer and mTOR inhibitors in solid tumor targeting therapy. Int J Mol Sci. 2019;20(3):755. https://doi.org/10.3390/ijms20030755.
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–6. https://doi.org/10.1158/0008-5472.CAN-09-1947.
Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013;110(29):11988–93. https://doi.org/10.1073/pnas.1310230110.
Li F, MacKenzie KR, Jain P, Santini C, Young DW, Matzuk MM. Metabolism of JQ1, an inhibitor of bromodomain and extra terminal bromodomain proteins, in human and mouse liver microsomes. Biol Reprod. 2020;103(2):427–36. https://doi.org/10.1093/biolre/ioaa043.
Zimmerman MW, Liu Y, He S, Durbin AD, Abraham BJ, Easton J, et al. MYC drives a subset of high-risk pediatric Neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. 2018;8(3):320–35. https://doi.org/10.1158/2159-8290.CD-17-0993.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8. https://doi.org/10.1038/s41392-020-0110-5.
Yoshida GJ. Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res. 2018;37(1):173. https://doi.org/10.1186/s13046-018-0835-y.
Rickman DS, Schulte JH, Eilers M. The expanding world of N-MYC-driven tumors. Cancer Discov. 2018 Feb;8(2):150–63. https://doi.org/10.1158/2159-8290.CD-17-0273.
Bahmad HF, Mouhieddine TH, Chalhoub RM, Assi S, Araji T, Chamaa F, et al. The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget. 2018;9(71):33549–61. https://doi.org/10.18632/oncotarget.26088.
Tahmasebi S, Amiri M, Sonenberg N. Translational control in stem cells. Front Genet. 2019;9:709. https://doi.org/10.3389/fgene.2018.00709.
Baser A, Skabkin M, Martin-Villalba A. Neural stem cell activation and the role of protein synthesis. Brain Plast. 2017;3(1):27–41. https://doi.org/10.3233/BPL-160038.
Kumari A, Folk WP, Sakamuro D. The dual roles of MYC in genomic instability and Cancer Chemoresistance. Genes (Basel). 2017;8(6):158. https://doi.org/10.3390/genes8060158.
Yogev O, Almeida GS, Barker KT, George SL, Kwok C, Campbell J, et al. In vivo modeling of Chemoresistant neuroblastoma provides new insights into Chemorefractory disease and metastasis. Cancer Res. 2019;79(20):5382–93. https://doi.org/10.1158/0008-5472.CAN-18-2759.
Jiang BH, Liu LZ. Role of mTOR in anticancer drug resistance: perspectives for improved drug treatment. Drug Resist Updat. 2008;11(3):63–76. https://doi.org/10.1016/j.drup.2008.03.001.
David-West G, Ernlund A, Gadi A, Schneider RJ. mTORC1/2 inhibition re-sensitizes platinum-resistant ovarian cancer by disrupting selective translation of DNA damage and survival mRNAs. Oncotarget. 2018;9(69):33064–76.
Reyes-González JM, Armaiz-Peña GN, Mangala LS, Valiyeva F, Ivan C, Pradeep S, et al. Targeting c-MYC in platinum-resistant ovarian Cancer. Mol Cancer Ther. 2015;14(10):2260–9. https://doi.org/10.1158/1535-7163.MCT-14-0801.
Miller AL, Fehling SC, Garcia PL, Gamblin TL, Council LN, van Waardenburg RCAM, et al. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine. 2019;44:419–30. https://doi.org/10.1016/j.ebiom.2019.05.035.
Geoerger B, Kieran MW, Grupp S, Perek D, Clancy J, Krygowski M, et al. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur J Cancer. 2012;48(2):253–62. https://doi.org/10.1016/j.ejca.2011.09.021.
Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol. 2017;18(7):946–57. https://doi.org/10.1016/S1470-2045(17)30355-8.
Acknowledgements
The authors thank the Flow Cytometry and Tissue Science Core Facilities at UNMC for their assistance with these studies. The authors thank Matt Sandbulte for his help in editing this manuscript. We thank all members of the CHRI-PCRG labs for their useful discussion and suggestion on this work.
Funding
This work was fully supported by the State of Nebraska through the Pediatric Cancer Research Grant Funds (LB905) awarded to D. W. Coulter, MD. This funding had no role in the study design, data collection and analysis, interpretation of the data, decision to publish, or writing the manuscript.
Author information
Authors and Affiliations
Contributions
NKC and DC designed the study. NKC, MJK, CNG, EMM, GA and SR performed the experiments and analyzed the data. KBC, SSJ and DC contributed significantly to the interpretation of the data. NKC and MJK wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kling, M.J., Griggs, C.N., McIntyre, E.M. et al. Synergistic efficacy of inhibiting MYCN and mTOR signaling against neuroblastoma. BMC Cancer 21, 1061 (2021). https://doi.org/10.1186/s12885-021-08782-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08782-9