
Abstract
Background
The molecular prognostic biomarkers of clear cell renal cell carcinoma (ccRCC) are still unknown. We aimed at researching the candidate biomarkers and potential therapeutic targets of ccRCC.
Methods
Three ccRCC expression microarray datasets (include GSE14762, GSE66270 and GSE53757) were downloaded from the gene expression omnibus (GEO) database. The differentially expressed genes (DEGs) between ccRCC and normal tissues were explored. The potential functions of identified DEGs were analyzed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). And then the protein - protein interaction network (PPI) was established to screen the hub genes. After that, the expressions of hub genes were identified by the oncomine database. The hub genes’ prognostic values of patients with ccRCC were analyzed by GEPIA database.
Results
A total of 137 DEGs were identified by utilizing the limma package and RRA method, including 63 upregulated genes and 74 downregulated genes. It is found that 137 DEGs were mainly enriched in 82 functional terms and 24 pathways in accordance with the research results. Thirteen highest-scoring genes were screened as hub genes (include 10 upregulated genes and 3 downregulated candidate genes) by utilizing the PPI network and module analysis. Through integrating the oncoming database and GEPIA database, the author found that C3 and CXCR4 are not only overexpressed in ccRCC, but also associated with the prognosis of ccRCC. Further results could reveal that patients with high C3 expression had a poor overall survival (OS), while patients with high CTSS and TLR3 expressions had a good OS; patients with high C3 and CXCR4 expressions had a poor disease-free survival (DFS), while ccRCC patients with high TLR3 expression had a good DFS.
Conclusion
These findings suggested that C3 and CXCR4 were the candidate biomarkers and potential therapeutic targets of ccRCC patients.
Similar content being viewed by others

Introduction
Renal cell carcinoma (RCC) is the most common kidney malignancies, which originates in the renal tubular epithelium [1]. Among of RCC, clear cell RCC (ccRCC) is the most important histological subtype, accounting for ∼80% of RCC [2]. The vast majority of RCC are discovered by accident. Less than 5% of RCC are detected by the classic triad (gross hematuria, flank pain and abdominal mass) and are often advanced. Due to resistant to radiotherapy and chemotherapy, surgical resection is still the optimal treatment for RCC [2]. Although the emergence of immunotherapy and targeted therapy has diversified the treatment of RCC, the prognosis of patients with RCC who have lost the opportunity of surgery remains dismal [3]. Therefore, it is particularly important to understand the pathogenesis of RCC and investigate biomarkers to support the treatment and prediction of prognosis.
In recent years, bioinformatics analysis of gene expression microarrays could help identify the potential target genes of diseases and provide the molecular characteristics, regulatory pathways and cellular networks of diseases [4]. The gene expression omnibus (GEO, www.ncbi.nlm.nih.gov/geo/) database is an international public functional genomics database, which stores common array and sequence data. In the past decades, more and more scholars had indicated that tumor-related genes were discovered by using GEO databases in their researches. For instance, Guo et al. found that 31 mostly changed hub genes were significant enriched in several pathways through integrated bioinformatical analysis, which mainly associated with cell cycle process, chemokines and G-protein coupled receptor signaling pathways in colorectal cancer [5]. Besides, Liang’s research results indicated that BCL2, CCND1 and COL1A1 might be the key genes in thyroid papillary carcinoma through bioinformatics analysis [6]. What’s more, bioinformatics has been widely used in the diagnosis and prognosis of renal cancer. For example, li et al. found that MMP2, DCN, COL4A1, CASR, GPR4, UTS2, and LDLR can be regarded as potential immunotherapy biomarkers for RCC [3]. And Tao constructed a immune-related gene-based prognostic index, which can effectively predict the prognosis of patients with renal cancer and the associated immunoinfiltrating cells and provide a new method for predicting the prognosis and targeted therapy of renal cancer [7].
Based on the above researches and methods, the author analyzed the gene expression profile of ccRCC by using the GEO database, and then further analyzed the data to provide valuable hub genes for the following translational and clinical research.
Materials and methods
Access to public resources
Three expression profiling datasets (GSE14762 [8], GSE66270 [9] and GSE53757 [10]) were downloaded from the Gene Expression Omni - bus (GEO) database of the National Center for Biotechnology Information (NCBI). The GSE14762 dataset included 11 tumor tissue samples and matched normal tissue samples. The GSE66270 dataset included 14 normal tissue samples and 14 tumor tissue samples. And the GSE53757 dataset included 72 tumor tissue samples and adjacent tissue samples. Among of them, the microarray data of GSE14762 was running at the GPL4866 Plaforms, and the microarray data of GSE66270 and GSE53757 were analyzed at the GPL570 Plaforms. Platforms and series matrix files were downloaded as TXT files. Details for GEO ccRCC data were shown in Table 1.
Detection of DEGs
The R language software (version 3.5.0; https://www.r-project.org/) and annotation package were used to handle the downloaded data files. Probe name in the downloaded data files was changed into international standard name. The package in the Bioconductor (http://www.bioconductor.org/) was used for gene distinguish expression analysis. Robust Multi-array Average (RMA) algorithm was used for the gene expression profile data preprocessing. And quantile normalization was performed to normalize the above data. P < 0.05 and [log2 Fold Change] ≥ 2 were regarded as the DEGs screening threshold. The Robust Rank Aggreg (RRA) analysis (http://cran.r-project.org/) was used to list the up-regulated and down-regulated genes. DEGs of three datasets were represented by volcano map and hierarchical clustering heat map.
Gene ontology (GO) and KEGG enrichment analyses
The biological processes (BP), molecular functions (MF) and cellular components (CC) of DEGs were explored by applying two online biological tools. The online website g:Profiler (https://biit.cs.ut.ee/gprofiler/gost) was used for Go analysis. And DAVID 6.8 (https://david.ncifcrf.gov/) was used for KEGG analysis. P < 0.05 was considered as the significant threshold for GO and KEGG pathway analysis.
PPI network construction
Online database STRING (http://string-db.org) and Cytoscape software (Version 3.6.1, http://www.cytoscape.org/) were applied to generate the PPI network of DEGs and identify the hub genes. Besides, the Molecular Complex Detection (MCODE) plug-in in Cytoscape software was used to analyze clustered sub-networks of highly intraconnected nodes from the above PPI network. The default parameters of MCODE plug-in were as follows: Degree cutoff ≥2, Node score cutoff ≥0.2, K-core ≥2, and Max depth = 100.
Expression and survival analysis of hub genes
The meta-analysis function of oncomine database (https://www.oncomine.org/) was used to better validate the expression level of hub genes. Besides, online database GEPIA (http://gepia.cancer-pku.cn/detail.php) was an interactive web server, which can analyze the expression of tumor and normal genes. The purpose of this study was to analyze the relationship between the hub genes expression and the survival analysis of [overall survival (OS) and disease free survival (DFS)].
Results
Microarray data information
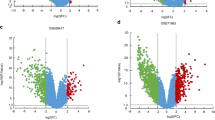
The RCC expression microarray datasets (GSE14762, GSE66270 and GSE53757) were standardized by RMA algorithm, and the results were shown in Fig. 1. The author obtained 381 DREs from GSE14762 in accordance with the screening criteria (P < 0.05 and [log2 FC] ≥ 2). Moreover, the author obtained 870 DEGs and 1324 DEGs from GSE66270 and GSE53757. The DEGs from the two groups of sample data included in each of the three databases were shown by volcano plot (Fig. 2). The cluster heatmaps of the top 100 DEGs from the three microarrays were shown in Fig. 3.

Standardization of gene expression by boxplot. The GSE14762 data (A), GSE66270 data (B) and GSE55757 data (C) was standardized

Volcano plot of differential data expressions between two sample sets. Three figures show the volcano plot of GSE14762 data (A), GSE66270 data (B) and GSE55757 data (C). The red oints represent overexpressed genes (threshold: P < 0.05 and |[log2 FC]| ≥ 2). The green points represent under-expressed genes (threshold: P < 0.05 and |[log2 FC]| ≥ 2). The black points represent undifferentiated genes

Clustering heatmap of DEGs. Three figures show the heatmap of GSE14762 data (A), GSE66270 data (B) and GSE55757 data (C). Red grid shows that the genes expression is uoverexpressed, green grid shows that the genes expression is under-expressed, black grid shows that there are no significant difference and gray grid shows that genes are too weak to be detected
DEGs identification in ccRCC
The three microarray databases of RCC were analyzed and sorted by the limma package (threshold: P < 0.05 and [log2 Fold Change] ≥ 2), and then further analyzed by the RRA method. As a result, 137 DEGs were identified, including 63 overexpressed genes and 74 under-expressed genes (Table 2). The heatmap of the top 20 overexpressed and under-expressed genes was revealed by R-heatmap software in Fig. 4.

RRA analyses. This figure shows the top 20 overexpressed and under-expressed genes obtained by RRA analyses. Red grid indicates that the genes expression is upregulated, blue grid indicates that the genes expression is downregulated, and white grid indicates that there is no detected gene expression
GO and KEGG analysis of DEG
The author futher understood the function of hub genes include BP, CC and MF by using DAVID database. Significant results of the GO enrichment analysis of DEGs in ccRCC are shown in Table 3. As shown in Fig. 5a and b, GO analysis (threshold: P < 0.05 and count≥2) demonstrated that ccRCC hub genes were mainly enriched in 50 terms of BP group, such as response to hypoxia, oxidation-reduction process and proteolysis. In CC group, DEGs were enriched in 21 terms, such as extracellular exosome, plasma membrane and membrane integral component. Similarly in MF group, DEGs were enriched in 11 terms, such as identical protein binding, receptor binding and heparin binding. As shown in Fig. 5c, the result illustrated the relationship between the different functions of cytoscape software.

Go and KEGG analysis of DREs in ccRCC. A DEGs were divided into three functional groups by GO analysis, including molecular function, biological processes and cell composition. B GO enrichment significance items of DEGs in three functional groups. C The relationship between the different functions. D Significant pathway enrichment of DEGs. Purple represents the signaling pathway, red represents the overexpressed genes and green represents the under-expressed genes
The significantly enriched pathways were submitted to KEGG analysis to further analyze the above DEGs. As shown in Table 4 and Fig. 5d, the significant pathway enrichment of DEGs was indicated by KEGG analysis. These DEGs were enriched in 24 pathways, which mainly related to metabolic pathways, phagosome and other pathways.
PPI network and module analysis
String database was used to generate PPI networks of DEGs in RCC. Figure 6a showed the relationship between the 137 candidate hub genes. Besides, MCODE application was applied to screen out the highest-scoring nodes. And Fig. 6b displayed the module with the highest score (score = 10, node = 11, edges = 50). As a result, MCODE application selected 13 nodes with the highest score, including 10 upregulated candidate genes (C1QA, C1QB, C3, CTSS, CXCR4, FCER1G, ITGB2, TLR2, TLR3 and TYROBP) and 3 downregulated candidate genes (AQP2, KNG1, PLG).

Protein-protein interaction network and MCODE application. A PPI network. B Top 13 degree genes by MCODE application
Expression and survival analysis of hub genes
The oncomine database and GEPIA database were applied to further explore the expression and prognosis of the above screened genes. Six analyses were obtained from the oncomine database (Fig. 7). The significant (P < 0.05) expression of 10 genes were suggested by the result of meta-analysis. Figure 8 indicated the OS and DFS of 10 genes. And the result demonstrated that ccRCC patients with high C3 expression had a poor OS, while ccRCC patients with high CTSS and TLR3 expressions had a good OS. Besides, in ccRCC patients, high C3 and CXCR4 expressions indicated a poor DFS, while high TLR3 expression indicated a good DFS. Finally, C3 and CXCR4 were selected to distinguish the prognosis of ccRCC patients.

The expression level of 13 hub genes. Among 6 different analysis datasets by the ONCOMINE database

The OS and DFS of 10 candidate genes in ccRCC patients by GEPIA database. (OS: A-J, DFS: K-T)
Discussion
Kidney cancer accounts for about 2 to 3% of adult malignant tumors, and 80 to 90% of adult renal malignancies. In 2012, about 338, 000 kidney cancer cases were newly discovered, accounting for 24% of all tumors; and there were 144,000 death cases, accounting for 17% of all tumors [11]. RCC was the most common kidney malignancies. The early symptoms of RCC were not obvious, and most patients are diagnosed with advanced stage or metastasis [12]. RCC was characteristic of easy recurrence and metastasis because of its complexity of the causes and pathogenesis. Moreover, it was insensitive to the traditional chemoradiotherapy. Under the influence of these reasons, RCC usually leaded to poor clinical outcomes. Hence, it could improve the diagnosis, treatment and prognosis of RCC via understanding more of the biological molecular mechanism.
The sequencing technology and bioinformatics are developing gradually, the collection and analysis of previous data will support to explor the pathogenesis of RCC and discover possible biomarkers for diagnosis andtreatment [13].
Bioinformatics method is a highly efficient research pathway, which could promote the development of related gene or group of disease by analyzing the biological data. At the present, Bioinformatics have been widely used at all areas, including medical research, the design of the discover disease-related genes, clinical diagnosis of disease, individualized treatment of diseases and new molecular targets for drug discovery [14].
One hundred thirty-seven DEGs were identified in this study, including 63 overexpressed genes and 74 under-expressed genes. It was found that these DEGs were mainly enriched in 82 terms and 24 pathways through GO and KEGG analysis. Thirteen highest-scoring genes were screened as hub gene through PPI network. Further verification based on the oncomine platform indicated that 10 hub genes (C1QA, C1QB, C3, CTSS, CXCR4, FCER1G, ITGB2, TLR2, TLR3 and TYROBP) had significantly highly expressed. Finally, through the GEPIA platform, the author found that ccRCC patients with high C3 expression had a poor OS, while ccRCC patients with high CTSS and TLR3 expressions had a better OS. Meanwhile, high C3 and CXCR4 expressions were associated with a poor DFS, while patients with high TLR3 expression had a good DFS.
As a protein coding gene, complement component 3 (C3) is involved in the occurrence and development of many diseases, including C3 deficiency, Autosomal Recessive and Hemolytic Uremic Syndrome, Atypical 5 [15]. And its related pathways are Immune response Lectin induced complement pathway and Signaling by GPCR. In previous reports, C3 was demonstrated as a potential prognostic marker for non-small cell lung cancer and may be a new immune marker to differentiate the prognosis of patients with non-small cell lung cancer [16, 17]. Besides, Yuan et al. demonstated that overexpressed C3 could activate the JAK2/STAT3 pathway, which affected the progression of gastric cancer [18]. In addition, it had been reported that tumor cell–derived C3 could regulated TAMs through C3a-C3aR-PI3Kγ pathway to suppress the antitumor immunity [19].
CTSS (Cathepsin S) is a protein coding gene. Previous articles in papillary thyroid carcinoma reported that CTSS was highly expressed and related to transformation. These results revealed that the highly expression of CTSS was associated with poor prognosis and lymph node metastasis [20]. Similarly, it had been reported that CTSS was over-expressed in triple-negative breast cancer, and the inhibition of CTSS could be conducted by inhibiting the growth and metastasis of triple-negative breast cancer [21]. Prof. Dheilly found that follicular lymphoma patients harbor a recurrent hotspot mutation targeting tyrosine 132 (Y132D) in cathepsin S (CTSS) that enhances protein activity. Futher study revealed that it could enhanced the anti-tumor immune responses in Non-Hodgkin Lymphoma by inhibiting CTSS [22]. In this study, the author analyzed the research data and found that CTSS was indeed highly expressed in RCC, but the high expression was associated with better prognosis. The prognosis of patients with high expression was even better, which is an opposite effect between expression and prognosis. The potential reasons for the inconsistent findings need further investigations.
As a member of the Toll-like receptor (TLR) family, previous studies had reported that TLR3 was abnormally expressed in a variety of tumors, including breast, ovarian and prostate tumors. But TLR3 was associated with the clinical outcomes of various cancers [23, 24]. Francesca revealed that TLR3 could induce apoptpsis in Non-Small-Cell Lung Cancer via boosting the innate immune response [25]. Besides, Fan’s result demonstated that TLR3 suppressed the proliferation by downregulating the EGFR/PI3K/AKT pathway in brest cancer [26]. Similarly, TLR3 was also downregulated in hepatocellular carcinoma. And deep reseach showed that overexpression of TLR3 was associated with longer survival [27]. In this study, TLR3 was highly expressed in RCC but it was related to the better prognosis result.
Chemokine receptor-4 (CXCR4) belongs to the super-family of the seven-transmembrane domain, heterotrimeric G-protein-coupled receptors and is associated with cell proliferation, migration, invasion and survival. In the previous reports, it had been demonstrated that CXCR4 was upregulated in sporadic Vestibular schwannomas (VS) as well as in neurofibromatosis type 2 (NF2) tumors [28,29,30]. Besides, SDF-1 (CXCL12)/CXCR4 signaling has been verified to play a vital role in oncobiology, especially in hypoxia adaptation, metastasis and migration [31]. What’s more, the CXCR4 antagonists (such as AMD3100, Mozobil®) were widely applied in hematopoietic stem cells, which could dramaticly increase the mobilization efficiency and yields of progenitor cells [32]. The results in this study showed that CXCR4 was over-expressed in RCC and associated with poor prognosis. However, the role of CXCR4 in RCC has been poorly studied. Therefore, the further exploration of the mechanism of CXCR4 in RCC will help people to find new therapeutic targets.
Conclusion
In summary, the author identified two ccRCC-associated candidate genes (C3 and CXCR4) with potential prognostic value via bioinformatics analysis of three expression profile datasets from the GEO database. Additionally, in this study, it have been found that CTSS and TLR3 were abnormally expressed in ccRCC and associated with ccRCC prognosis. However, their expression level is contrary to the prognosis. These novel biomarkers may have important clinical significance for the diagnosis and prognosis of RCC, but their detailed action mechanism in the development of renal carcinoma needs to be further explored. In the following studies, the author will further verify the expression of the above genes in renal cancer through RT-QPCT. In addition, its downstream target genes and signaling pathways need to be explored and verified by cell experiments in vitro and animal experiments in vivo, which will help the author to better understand its developmental mechanism in renal cancer.
Availability of data and materials
The data used to support the findings of this study are all included within the thesis. The gene expression data can be accessed on Gene Expression Omnibus (GEO). Oncomine database was used to get the expression profile of hub genes. GEPIA database was used to obtain the overall survival and disease free survival analysis of genes.
The datasets analysed during this study are available in the:
1. GSE14762_RAW: https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE14762&format=file
2. GSE15641_RAW: https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE15641&format=file
3. GSE53757_RAW: https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE53757&format=file
4. GSE53757_series_matrix.txt: https://ftp.ncbi.nlm.nih.gov/geo/series/GSE53nnn/GSE53757/matrix/GSE53757_series_matrix.txt.gz
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. https://doi.org/10.3322/caac.21442.
Escudier B, Porta C, Schmidinger M, Rioux-Leclercq N, Bex A, Khoo V, et al. Renal cell carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann Oncol. 2019;30(5):706–20. https://doi.org/10.1093/annonc/mdz056.
Li F, Guo P, Dong K, Guo P, Wang H, Lv X. Identification of key biomarkers and potential molecular mechanisms in renal cell carcinoma by bioinformatics analysis. J Comput Biol. 2019;26(11):1278–95. https://doi.org/10.1089/cmb.2019.0145.
Alizadeh M, Valizadeh R, Rahimi MM. Immediate successful renal autotransplantation after proximal ureteral avulsion fallowing ureteroscopy: a case report. J Surg Case Rep. 2017;2017(2).
Guo Y, Bao Y, Ma M, Yang W. Identification of key candidate genes and pathways in colorectal cancer by integrated bioinformatical analysis. Int J Mol Sci. 2017;18(4).
Liang W, Sun F. Identification of key genes of papillary thyroid cancer using integrated bioinformatics analysis. J Endocrinol Investig. 2018;41(10):1237–45. https://doi.org/10.1007/s40618-018-0859-3.
Tao H, Li Z, Mei Y, Li X, Lou H, Dong L, et al. Integrative bioinformatics analysis of a prognostic index and immunotherapeutic targets in renal cell carcinoma. Int Immunopharmacol. 2020;87:106832. https://doi.org/10.1016/j.intimp.2020.106832.
Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med. 2009;15(3):319–24. https://doi.org/10.1038/nm.1922.
Wotschofsky Z, Gummlich L, Liep J, Stephan C, Kilic E, Jung K, et al. Integrated microRNA and mRNA signature associated with the transition from the locally confined to the metastasized clear cell renal cell carcinoma exemplified by miR-146-5p. PLoS One. 2016;11(2):e0148746. https://doi.org/10.1371/journal.pone.0148746.
von Roemeling CA, Radisky DC, Marlow LA, Cooper SJ, Grebe SK, Anastasiadis PZ, et al. Neuronal pentraxin 2 supports clear cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-4. Cancer Res. 2014;74(17):4796–810. https://doi.org/10.1158/0008-5472.CAN-14-0210.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. https://doi.org/10.1002/ijc.29210.
Desar IM, Jacobs JH, Hulsbergen-vandeKaa CA, Oyen WJ, Mulders PF, van der Graaf WT, et al. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int J Cancer. 2011;129(2):507–12. https://doi.org/10.1002/ijc.25674.
Lyu J, Zhu Y, Zhang Q. An increased level of MiR-222-3p is associated with TMP2 suppression, ERK activation and is associated with metastasis and a poor prognosis in renal clear cell carcinoma. Cancer Biomark. 2020;28(2):141–9. https://doi.org/10.3233/CBM-190264.
Cheng L, Schneider BP, Li L. A bioinformatics approach for precision medicine off-label drug drug selection among triple negative breast cancer patients. J Am Med Inform Assoc. 2016;23(4):741–9. https://doi.org/10.1093/jamia/ocw004.
Assirelli E, Pulsatelli L, Dolzani P, Mariani E, Lisignoli G, Addimanda O, et al. Complement expression and activation in osteoarthritis joint compartments. Front Immunol. 2020;11:535010. https://doi.org/10.3389/fimmu.2020.535010.
Xu J, Nie H, He J, Wang X, Liao K, Tu L, et al. Using machine learning modeling to explore new immune-related prognostic markers in non-small cell lung Cancer. Front Oncol. 2020;10:550002. https://doi.org/10.3389/fonc.2020.550002.
Fayad C, Audi H, Khnayzer RS, Daher CF. The anti-cancer effect of series of strained photoactivatable Ru(II) polypyridyl complexes on non-small-cell lung cancer and triple negative breast cancer cells. J Biol Inorg Chem. 2021;26(1):43–55. https://doi.org/10.1007/s00775-020-01835-7.
Yuan K, Ye J, Liu Z, Ren Y, He W, Xu J, et al. Complement C3 overexpression activates JAK2/STAT3 pathway and correlates with gastric cancer progression. J Exp Clin Cancer Res. 2020;39(1):9. https://doi.org/10.1186/s13046-019-1514-3.
Zha H, Wang X, Zhu Y, Chen D, Han X, Yang F, et al. Intracellular activation of complement C3 leads to PD-L1 antibody treatment resistance by modulating tumor-associated macrophages. Cancer Immunol Res. 2019;7(2):193–207. https://doi.org/10.1158/2326-6066.CIR-18-0272.
Tan J, Qian X, Song B, An X, Cai T, Zuo Z, et al. Integrated bioinformatics analysis reveals that the expression of cathepsin S is associated with lymph node metastasis and poor prognosis in papillary thyroid cancer. Oncol Rep. 2018;40(1):111–22. https://doi.org/10.3892/or.2018.6428.
Gautam J, Banskota S, Lee H, Lee YJ, Jeon YH, Kim JA, et al. Down-regulation of cathepsin S and matrix metalloproteinase-9 via Src, a non-receptor tyrosine kinase, suppresses triple-negative breast cancer growth and metastasis. Exp Mol Med. 2018;50(9):1–14. https://doi.org/10.1038/s12276-018-0135-9.
Dheilly E, Battistello E, Katanayeva N, Sungalee S, Michaux J, Duns G, et al. Cathepsin S regulates antigen processing and t cell activity in non-hodgkin lymphoma. Cancer Cell. 2020;37(5):674–689.e612.
Huang Y, Li X, Pan C, Cheng W, Wang X, Yang Z, et al. The intervention mechanism of emodin on TLR3 pathway in the process of central nervous system injury caused by herpes virus infection. Neurol Res. 2021;43(4):307–13. https://doi.org/10.1080/01616412.2020.1853989.
Muresan XM, Bouchal J, Culig Z, Souček K. Toll-Like Receptor 3 in solid cancer and therapy resistance. Cancers (Basel). 2020;12(11).
Bianchi F, Alexiadis S, Camisaschi C, Truini M, Centonze G, Milione M, et al. TLR3 expression induces apoptosis in human non-small-cell lung cancer. Int J Mol Sci. 2020;21(4).
Fan L, Zhou P, Hong Q, Chen AX, Liu GY, Yu KD, et al. Toll-like receptor 3 acts as a suppressor gene in breast cancer initiation and progression: a two-stage association study and functional investigation. Oncoimmunology. 2019;8(6):e1593801. https://doi.org/10.1080/2162402X.2019.1593801.
Yuan MM, Xu YY, Chen L, Li XY, Qin J, Shen Y. TLR3 expression correlates with apoptosis, proliferation and angiogenesis in hepatocellular carcinoma and predicts prognosis. BMC Cancer. 2015;15(1):245. https://doi.org/10.1186/s12885-015-1262-5.
Fan G, Wei J. Identification of potential novel biomarkers and therapeutic targets involved in human atrial fibrillation based on bioinformatics analysis. Kardiol Pol. 2020;78(7–8):694–702. https://doi.org/10.33963/KP.15339.
Lassaletta L, Calvino M, Morales-Puebla JM, Lapunzina P, Rodriguez-de la Rosa L, Varela-Nieto I, et al. Biomarkers in vestibular schwannoma-associated hearing loss. Front Neurol. 2019;10:978.
Chifu I, Heinze B, Fuss CT, Lang K, Kroiss M, Kircher S, et al. Impact of the chemokine receptors CXCR4 and CXCR7 on clinical outcome in adrenocortical carcinoma. Front Endocrinol (Lausanne). 2020;11:597878. https://doi.org/10.3389/fendo.2020.597878.
Eckert F, Schilbach K, Klumpp L, Bardoscia L, Sezgin EC, Schwab M, et al. Potential role of CXCR4 targeting in the context of radiotherapy and immunotherapy of cancer. Front Immunol. 2018;9:3018. https://doi.org/10.3389/fimmu.2018.03018.
Devine SM, Vij R, Rettig M, Todt L, McGlauchlen K, Fisher N, et al. Rapid mobilization of functional donor hematopoietic cells without G-CSF using AMD3100, an antagonist of the CXCR4/SDF-1 interaction. Blood. 2008;112(4):990–8. https://doi.org/10.1182/blood-2007-12-130179.
Acknowledgements
Not applicable.
Funding
This study was supported by the General Project Funds from the Health Department of Zhejiang Province (Grant no 2021439557).
Author information
Authors and Affiliations
Contributions
Protocol/project development—DaHong Zhang; Data collection or management JingQuan,Yuchen Bai, and ErLei Han; Data analysis—YunBei Yang,Hong Bai and Qi Zhang; Manuscript writing/editing—YuChen Bai and Jing Quan; The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Zhejiang Provincial Peoples’ Hospital, Peoples’ Hospital of Hangzhou Medical College. Ethics approval was not required to access the data in the first instance. Ethical approval provided the permission to access, analyze and publish all the data obtained.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Quan, J., Bai, Y., Yang, Y. et al. Bioinformatics analysis of C3 and CXCR4 demonstrates their potential as prognostic biomarkers in clear cell renal cell carcinoma (ccRCC). BMC Cancer 21, 814 (2021). https://doi.org/10.1186/s12885-021-08525-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08525-w