
Abstract
Background
The chemotherapy triplet FOLFOXIRI combined to the anti-VEGF antibody bevacizumab is an option in selected patients with metastatic colorectal cancer. In this setting, RAS-mutated metastatic colorectal cancer do not benefit the same from treatment than RAS-wildtype metastatic colorectal cancer do. Together with its antiangiogenic properties, the tyrosine-kinase inhibitor regorafenib has also anti-proliferative activities whatever the RAS status is. The present trial aims at studying the safety and the efficacy of regorafenib in combination with FOLFIRINOX – a chemotherapy triplet using a different dosing schedule than FOLFOXIRI - in patients with RAS-mutated metastatic colorectal cancer.
Methods
FOLFIRINOX-R is a prospective, multicentric, non-randomised, dose-finding phase 1–2 trial. The primary endpoints are the determination of the maximum tolerated dose, the recommended phase 2 dose, and the proportion of patients achieving disease control at 48-weeks. Phase 1 follows a 3 + 3 design (12 to 24 patients to be included). Sixty nine patients will be necessary in phase 2, including 5% non-evaluable ones, with the following assumptions, one-stage Fleming design, α = 5%, β = 20%, p0 = 35% and p1 = 50%. Key eligibility criteria include Eastern Cooperative Oncology Group Performance Status of ≤1 and RAS-mutated metastatic colorectal cancer not amenable to surgery with curative intent and not previously treated for metastatic disease. FOLFIRINOX (oxaliplatin 85 mg/m2, folinic acid 400 mg/m2, irinotecan 150–180 mg/m2, 5-fluorouracil: 400 mg/m2 then 2400 mg/m2 over 46 h) is administered every 14 days. Regorafenib (80 to 160 mg, as per dose-level) is administered orally, once daily on days 4 to 10 of each cycle.
Discussion
FOLFIRINOX-R is the first phase I/II study to evaluate the safety and efficacy of regorafenib in combination with FOLFIRINOX as frontline therapy for patients with RAS-mutated metastatic colorectal cancer.
Trial registration
EudraCT: 2018-003541-42; ClinicalTrials.gov: NCT03828799.
Similar content being viewed by others


Background
Metastatic colorectal cancer
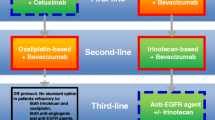
Colorectal cancer (CRC) is a major cause of morbidity and mortality globally [1]. More than 50% of patients will develop metastatic disease, and while some patients with technically resectable liver (or lung) metastases could be cured with surgery only, most of the patients will require palliative systemic therapy as their metastases are found unresectable [2]. As per European Society for Medical Oncology guidelines, frontline chemotherapy for metastatic CRC (mCRC) commonly involves the doublet (2-CTx) regimens of 5-fluorouracil, folinic acid, and either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) [2]. However, Falcone et al. [3] reported higher response rates and better survival rates with the use of a chemotherapy triplet (3-CTx) that combined 5-fluorouracil, folinic acid, oxaliplatin and irinotecan (FOLFOXIRI) over 2-CTx. Similarly, our group reported favorable outcomes in patients with unresectable liver metastases from colorectal origin, with the same three drugs but using a different dosing schedule, i.e. the FOLFIRINOX regimen [4]. It is also accepted that the addition of targeted therapies such as bevacizumab (a monoclonal antibody which binds circulating vascular endothelial growth factor-A) or as monoclonal antibodies (cetuximab, panitumumab) directed to epidermal growth factor receptor to 2-CTx or 3-CTx it is beneficial in fit patients when tumor shrinkage is a major objective to allow conversion surgery [2].
RAS mutations are identified in about 60% of mCRC tumors and are known as negative predictors of efficacy for anti-epidermal growth factor receptor therapy in mCRC [5]. RAS status also carries distinct prognostic information, as reported in a series of 1239 mCRC patients who have been treated with 2-CTx in five randomized trials [5]. Interestingly, the poor prognosis related to RAS mutations was observed across different treatment regimens (subgroups of irinotecan- and oxaliplatin-treated patients as well as in bevacizumab- and non-bevacizumab-treated patients). Median progression-free survival (PFS) and overall survival (OS) were 10.3 vs. 9.5 months and 26.9 vs. 21.1 months in RAS-wild type (and BRAF-wild type) and RAS-mutated tumors, respectively [5]. Similar survival differences by RAS status were also observed in clinical trials which addressed the benefit of 3-CTx over 2-CTx. The TRIBE consortium reported that 3-CTx (FOLFOXIRI) combined with bevacizumab provided a significantly longer PFS and OS than did 2-CTx with FOLFIRI plus bevacizumab [6, 7]. Actually, when looking at survival by RAS status, Cremolini et al. [7] reported that median OS was 37.1 months (95%CI, 29.7–42.7) in the RAS- and BRAF-wild-type subgroup compared with 25.6 months (95%CI, 22.4–28.6) in the RAS-mutated subgroup (HR 1.49, 95%CI, 1.11–1·99). For sure, we need to increase the treatment benefit in this setting of RAS-mutated mCRC.
Regorafenib
Regorafenib is a small-molecule inhibitor of multiple membrane-bound and intracellular kinases such as RET, VEGFR-1, − 2, − 3, c-KIT, PDGFR, FGFR1, TIE2, DDR1, Trk2A, Eph2A, SAPK2, PTK5, Abl, CSF1R, RAF-1, BRAF, and BRAF V600E [8] which is approved for refractory mCRC. Beyond its well-known antiangiogenic properties, regorafenib has also less-known anti-proliferative activities in human colon cancer cell lines [8]. Interestingly, regorafenib potently inhibits growth of patient-derived CRC xenografts alone and in combination with irinotecan [8,9,10]. Two phase III trials demonstrated an overall survival benefit for regorafenib (160 mg orally, once daily for the first 21 days of each 28-day cycles) over placebo in patients with mCRC who progressed on standard therapies, whatever the RAS status was [11, 12]. However, half of the patients who received regorafenib in that setting progressed 2 months after the onset of treatment [11, 12] and were then possibly unnecessary exposed to adverse events (AEs). Unfortunately, there is no fully accepted biomarker able to predict regorafenib benefit. Two phase II trials also studied the safety and efficacy profile of regorafenib when combined to chemotherapy in patients with mCRC [13, 14]. The study from Schultheis et al. [13] was designed to explore whether the addition of regorafenib to FOLFOX or FOLFIRI could be feasible as first- or second-line treatment of mCRC. Forty-five patients were treated every 2 weeks with 5-fluorouracil 400 mg/m2 bolus then 2400 mg/m2 over 46 h, folinic acid 400 mg/m2, and either oxaliplatin 85 mg/m2 or irinotecan 180 mg/m2. On days 4 to 10, patients received regorafenib 160 mg orally once daily. Regorafenib showed acceptable tolerability in combination with chemotherapy. The most frequent grade 3–4 AEs were: neutropenia (45%), Hand-Foot Skin Reaction (15%), diarrhea (10%), and hypophosphatemia (12%). The study from Sanoff et al. [14] was designed to show whether the addition of regorafenib to FOLFIRI (same regimen than in the Schultheis trial [13]) improves PFS (over a placebo-FOLFIRI arm) when given as second-line treatment for patients previously treated with oxaliplatin and fluoropyrimidine-based regimen. The study met its primary endpoint, demonstrating that the addition of regorafenib to FOLFIRI prolongs PFS compared to FOLFIRI alone with a HR of 0.72. When looking at tumor response, the authors found that regorafenib (combined to chemotherapy) provided more partial responses than chemotherapy plus placebo (35% vs. 19%, p = 0.045). Tolerability was acceptable, with little increase in toxicity compared to the control chemotherapy regimen [14]. Based on these encouraging results, it could be worth studying the combination of 3-CTx to regorafenib, with the ultimate objective of providing a more efficacious alternative to a selected population of eligible patients.
Aim of the study
The present FOLFIRINOX-R trial aims at studying the safety, tolerability and efficacy of regorafenib in combination with FOLFIRINOX in patients with RAS-mutated mCRC.
Methods and design
Study design and treatment
FOLFIRINOX-R trial is a standard 3 + 3 design for dose escalation/de-escalation followed by a phase II trial when the recommended phase II dose (RP2D) is determined. It is designed as a single-arm, prospective, non-randomized, open label, multicenter, dose-finding phase I/II trial. It aims at determining the maximum tolerated dose (MTD) and the RP2D of the combination of regorafenib and FOLFIRINOX in the phase I-part of the trial. It aims too at evaluating the efficacy of the treatment in the phase II - part of the trial.
FOLFIRINOX is administered as per standard procedures every 14 days (1 cycle = 14 days) as follows, oxaliplatin 85 mg/m2 on day 1, IV infusion over 2 h, immediately followed by folinic acid 400 mg/m2 or calcium levofolinate 200 mg/m2 given as a 2-h IV infusion, with the addition of irinotecan 150–180 mg/m2 as per dose-level given as a 90-min intravenous infusion through a Y-connector immediately followed by 5-fluorouracil: 400 mg/m2 IV bolus then 2400 mg/m2 over 46 h continuous infusion. Primary prophylactic G-CSF is delivered from Day-7 to Day-12. Regorafenib is administered orally at a dose of 80 to 160 mg, as per dose-level, once daily on days 4 to 10 of each cycle (see Fig. 1 for an overview of the study design).

Treatment design
The pre-defined dose levels are the following, step − 1 (0 to 6 patients to be treated): irinotecan 150 mg/m2 plus regorafenib 80 mg/m2; step 1 (3 to 6 patients to be treated): irinotecan 150 mg/m2 plus regorafenib 120 mg/m2; step 2 (3 to 6 patients to be treated): irinotecan 180 mg/m2 plus regorafenib 120 mg/m2; step 3 (3 to 6 patients to be treated): irinotecan 180 mg/m2 plus regorafenib 160 mg/m2.
Twelve cycles of FOLFIRINOX have to be delivered consecutively or not. Indeed, treatment can be temporarily stopped to perform surgical resection or regional procedures (radiofrequency, cryoablation, radiation therapy) if the disease becomes accessible to them but if it happens, before the end of the 12 planned cycles, the treatment has to be resumed within 4 to 8 weeks after the loco-regional procedure completion in order to reach a total number of 12 cycles delivered. Regorafenib dosing is 80 to 160 mg/m2 according to each level of dose, by mouth daily on days 4 to 10 of each 14 days cycle, according with the regorafenib schedule reported by Schultheis et al. [13]. Regorafenib is continued until tumor progression or unacceptable toxicity, even after 12 cycles of FOLFIRINOX. Clinical evaluation will be made at each cycle. The following biologic assessments are performed before each cycle: RBC, hemoglobin, hematocrit, platelet count, WBC, coagulation panel (Prothrombin time, Partial Thromboplastin Time), electrolyte panel (sodium, potassium, chloride, magnesium, calcium, bicarbonates, phosphate), chemistry panel (albumin, lipase, glucose, serum creatinine, MDRD clearance, uric acid, urea, protein total), hepatic panel (AST, ALT, direct and indirect bilirubin, alkaline phosphatase, gammaglutamyl transpeptidase, lactate dehydrogenase), urine analysis (PH, protein, glucose, bilirubin, ketones, blood cells and leukocytes). TSH is performed every 6 weeks. The following biologic assessments is performed on day 8 of cycle 1, 2, 3: RBC, hemoglobin, hematocrit, platelet count and WBC (including differential neutrophil, lymphocyte, monocyte, basophil and eosinophil counts), chemistry panel (albumin, total protein, lipase, glucose, serum creatinine, MDRD clearance, uric acid and Urea), liver function panel (AST, ALT, direct and indirect bilirubin, gammaglutamyl transpeptidase, lactate dehydrogenase). Blood pressure is monitored every other week for the three first cycles of study treatment. Clinical, morphological imaging (RECIST criteria, v1.1) and biological (CEA and CA19.9) tumor assessment are done every 8 weeks. Prespecified dose adjustments (dose modifications and/or dose delay) for regorafenib or FOLFIRINOX could be made to manage adverse events.
Recruitment is currently active in three cancer centers (Dijon, Montpellier, and Nice) in France. Written informed consent is obtained from each patient before any screening and inclusion procedure. Patients remain on study until one of the following condition applies, study withdrawal, discontinuation of treatment or death.
Study objectives and endpoints
The primary objective of the phase 1 – part of the study is to determine the MTD and the RP2D of the combination of regorafenib and FOLFIRINOX. DLT is defined as the occurrence of one or more of the following toxicities during the three first cycles of treatment:
-
Unplanned interruption > 7 days of regorafenib due to drug-related toxicity
-
Grade (gr.) ≥ 3 (CTCAE v5) non-hematologic toxicity, except: gr. 3 nausea, gr. 3 vomiting, gr. 3 diarrhea, and gr. ≥ 3 lipase elevation without signs of pancreatitis,
-
Gr. ≥ 2 posterior reversible encephalopathy syndrome,
-
Gr. ≥ 2 retinopathy,
-
Any of the following liver-specific DLTs: gr. ≥ 3 bilirubin increase, gr. ≥ 3 AST and/or ALT increase, or AST and/or ALT increase > 3 x UNL with concurrent bilirubin increase (> 2 x UNL)
-
Gr. 4 neutropenia lasting > 3 days,
-
Gr. ≥3 febrile neutropenia (ANC < 1000/mm3 with fever ≥38.5 °C),
-
Gr. 4 anemia,
-
Platelets < 25,000 /mm3 or platelets < 50,000 /mm3 with bleeding,
-
INR or PTT elevation of Gr. ≥ 3 with bleeding,
-
Gr. ≥ 3 hemorrhage/bleeding events.
The primary objective of the Phase 2 – part of the study is to evaluate the efficacy, with the 48-week disease-control rate assessment. The latter is defined as the rate of non-progressing patients (after central review) at 48-week post-initiation of therapy, in all treated patients.
The secondary endpoints of the phase 2 – part of the project include safety (according to NCI-CTC v5 scale), overall response rate (according to RECIST v1.1), duration of response, 8-week response rate, deepness of response, disease-control rate, resectability (R0/R1) rate among patients with liver-only metastases, progression-free survival, progression-free survival since maintenance therapy onset with regorafenib, and the overall survival.
Statistical design
A minimum of 12 and a maximum of 24 patients will be included in the phase I – part of the study, with a minimum of 3 and a maximum of 6 patients per dose level. Six patients will be included at the RP2D. It would be necessary to include 65 evaluable patients (actually 69 patients, including 5% non-evaluable patients) in the phase II-part of the trial with the following assumptions, one-stage Fleming design, α = 5%, β = 20%, p0 (the probability of maximal inefficiency) = 35% and p1 (the probability of minimal efficiency) = 50%. FOLFIRINOX-R will be considered sufficiently effective (rejection of the null hypothesis) if at least 29 successes occur (defined as a 48-week disease control) out of 65 evaluable patients, the success rate (48-weeks disease control rate) being significantly greater than 35%. FOLFIRINOX-R will be considered insufficiently effective (rejection of the alternative hypothesis) in case of 28 or less successes out of 65 evaluable patients, the success rate (48-weeks disease control rate) being significantly lower than 50%. Overall, a maximum of 87 patients (18 patients for phase I + 69 patients for phase II, including 6 patients treated at the RP2D during the phase I) will be included.
Study procedures
Enrollment into FOLFIRINOX-R will be performed in two stages (a molecular testing then a full testing if appropriate) (Fig. 2) with two separate inform-consent forms. Only patients with the following characteristics, histological or cytological documented metastatic colorectal cancer not amenable to surgical resection with curative intent, no prior therapy for metastatic disease, measurable disease, aged 18 or older, with an Eastern Cooperative Oncology Group performance status ≤1 and a life expectancy of at least 3 months, will undergo the molecular testing which includes a circulating cell-free DNA (ccfDNA) RAS-mutation detection, a serum uracil assessment, and an assessment of the polymorphisms of UGT1A*28.

Selection procedure
Study population
The patients identified with all the conditions or characteristics listed Table 1 could be included in the study, whereas those identified with at least one condition or characteristic listed Table 2 could not be included.
Biomarker studies and translational analyses
Serial blood samples are collected at several time points: at baseline before study treatment, every 2 cycles of treatment, and at treatment discontinuation. We monitor ccfDNA for efficacy or resistance of study treatment. We also plan to look within the available colorectal samples at the activity and expression of selected kinases been targeted by regorafenib.
Ethics and regulatory considerations
The study was approved by French regulatory authorities (Agence nationale de sécurité du médicament et des produits de santé) on Jan 24th 2019, received a favorable opinion by the “Comité de Protection des Personnes EST III” (Dec 6th, 2018) and complies with the Helsinki declaration and French laws and regulations, and follows the International Conference on Harmonisation E6 (R2) Guideline for Good Clinical Practice. The trial results, even inconclusive, will be submitted in a peer-reviewed journal.
Discussion
In fit patients with primarily unresectable mCRC, it becomes increasingly obvious to use frontline 3-CTx alone or combined with a targeted-agent, to get enough tumor shrinkage to allow conversion surgery or significant control of tumor burden. In this setting, RAS-mutated mCRC do not benefit the same from treatment than RAS-wild type mCRC do. Therefore, it appears reasonable to combine 3-CTx to the antiangiogenic agent bevacizumab in RAS-mutated mCRC, with the hope of improving outcome [2]. Unlike what it is known with bevacizumab, regorafenib exhibits not only antiangiogenic properties with cytostatic effects but also true cytotoxic effects as demonstrated in about 3% of heavily pretreated mCRC patients who presented with a tumor response in the CORRECT study [11, 15]. It is also worth noting that a clinical benefit favoring regorafenib over placebo was identified across RAS status [11, 12]. As preliminary studies have reported the safety and the efficacy of combining FOLFOX or FOLFIRI with regorafenib in mCRC [13, 14], it seems to us that it could be worth studying the combination of 3-CTx to regorafenib in RAS-mutant mCRC patients. On that line, we expect that this FOLFIRINOX-regorafenib combination will be able to challenge the FOLFIRINOX-bevacizumab regimen in primarily unresectable mCRC patients with RAS-mutant tumors.
We preferentially used FOLFIRINOX as backbone 3-CTx rather than FOLFOXIRI which was investigated by the GONO group [6] because we have a pioneered and long-lasting experience with FOLFIRINOX, both in the metastatic and in the adjuvant setting in colorectal and pancreatic cancer [4, 16,17,18]. The key difference between FOLFIRINOX and FOLFOXIRI relies on irinotecan and fluorouracil dosing (FOLFIRINOX: irinotecan 150 mg/m2, fluorouracil 2400 mg/m2 as a continuous infusion plus bolus fluorouracil 400 mg/m2; FOLFOXIRI: irinotecan 165 mg/m2, fluorouracil 3200 mg/m2 as a continuous infusion without any bolus of fluorouracil). As there is no evidence that FOLFIRINOX is different from FOLFOXIRI, we use the 3-CTx regimen we are confident with, i.e.: the FOLFIRINOX regimen. We used regorafenib continuation beyond the scheduled interruption of chemotherapy after 12 cycles. For patients who achieved disease control after 4 cycles of chemotherapy (most of the time 2-CTx with or without bevacizumab), the most common maintenance strategies are the following: maintenance with fluoropyrimidine or with fluoropyrimidine plus bevacizumab or observation [19]. Less is known for patients achieving disease control with 12 cycles of 3-CTx plus bevacizumab. In this clinical trial, the regorafenib use is all the more relevant for maintenance therapy that it provides (as opposed to bevacizumab monotherapy) an already demonstrated antitumour activity in mCRC [11, 12].
Conclusion
The FOLFIRINOX-R study is the first phase I/II study to evaluate the safety and efficacy of regorafenib in combination with FOLFIRINOX as initial therapy for patients with primarily unresectable mCRC, which are found RAS-mutated on plasmatic ccfDNA assessment. Our objective is to demonstrate that this FOLFIRINOX-regorafenib combination provides enough clinical benefit to be tested in a controlled study in a selected patient population.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed up to now for the current study.
Abbreviations
- CRC:
-
Colorectal cancer
- mCRC:
-
Metastatic colorectal cancer
- 2-CTx:
-
Chemotherapy doublet
- 3-CTx:
-
Chemotherapy triplet
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- ccfDNA:
-
Circulating cell-free DNA
References
Bray F, Colombet M, Mery L, Piñeros M, Znaor A, Zanetti R, Ferlay J, editors. Cancer Incidence in Five Continents, Vol. XI. IARC Scientific Publication No. 166. Lyon: International Agency for Research on Cancer; 2020. Available from: https://publications.iarc.fr/597.
Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–422. https://doi.org/10.1093/annonc/mdw235.
Falcone A, Ricci S, Brunetti I, Pfanner E, Allegrini G, Barbara C, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer. J Clin Oncol. 2007;25(13):1670–6. https://doi.org/10.1200/JCO.2006.09.0928.
Ychou M, Rivoire M, Thezenas S, Quenet F, Delpero JR, Rebischung C, et al. A randomized phase II trial of three intensified chemotherapy regimens in first-line treatment of colorectal cancer patients with initially unresectable or not optimally resectable liver metastases. The METHEP trial. Ann Surg Oncol. 2013;20(13):4289–97. https://doi.org/10.1245/s10434-013-3217-x.
Modest DP, Ricard I, Heinemann V, Hegewisch-Becker S, Schmiegel W, Porschen R, et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol. 2016;27(9):1746–17. https://doi.org/10.1093/annonc/mdw261.
Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371(17):1609–18. https://doi.org/10.1056/NEJMoa1403108.
Cremolini C, Loupakis F, Antoniotti C, Lupi C, Sensi E, Lonardi S, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16(13):1306–15. https://doi.org/10.1016/S1470-2045(15)00122-9.
Schmieder R, Hoffmann J, Becker M, Bhargava A, Müller T, Kahmann N, et al. Regorafenib (BAY 73-4506): antitumor and antimetastatic activities in preclinical models of colorectal cancer. Int J Cancer. 2014;135(6):1487–96. https://doi.org/10.1002/ijc.28669.
Mross K, Frost A, Steinbild S, Hedbom S, Büchert M, Fasol U, et al. Phase I dose-escalation study of regorafenib (BAY 73 4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(9):2658–67. https://doi.org/10.1158/1078-0432.CCR-11-1900.
Jeong W, Doroshow JH, Kummar S. US FDA approved oral kinase inhibitors for the treatment of malignancies. Curr Probl Cancer. 2013;37(3):110–44. https://doi.org/10.1016/j.currproblcancer.2013.06.001.
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo controlled, phase 3 trial. Lancet. 2013;381(9863):303–12. https://doi.org/10.1016/S0140-6736(12)61900-X.
Li J, Qin S, Xu R, Yau TC, Ma B, Pan H, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015;16(6):619–29. https://doi.org/10.1016/S1470-2045(15)70156-7.
Schultheis B, Folprecht G, Kuhlmann J, Ehrenberg R, Hacker UT, Köhne CH, et al. Regorafenib in combination with FOLFOX or FOLFIRI as first- or second-line treatment of colorectal cancer: results of a multicenter phase Ib study. Ann Oncol. 2013;24(6):1560–7. https://doi.org/10.1093/annonc/mdt056.
Sanoff HK, Goldberg RM, Ivanova A, O'Reilly S, Kasbari SS, Kim RD, et al. Multicenter, randomized, double-blind phase 2 trial of FOLFIRI with regorafenib or placebo as second-line therapy for metastatic colorectal cancer. Cancer. 2018;124(15):3118–26. https://doi.org/10.1002/cncr.31552.
Tabernero J, Lenz HJ, Siena S, Sobrero A, Falcone A, Ychou M, et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015;16(8):937–48. https://doi.org/10.1016/S1470-2045(15)00138-2.
Ychou M, Viret F, Kramar A, Desseigne F, Mitry E, Guimbaud R, et al. Tritherapy with fluorouracil/leucovorin, irinotecan and oxaliplatin (FOLFIRINOX): a phase II study in colorectal cancer patients with non-resectable liver metastases. Cancer Chemother Pharmacol. 2008;62(2):195–201. https://doi.org/10.1007/s00280-007-0588-3.
Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25. https://doi.org/10.1056/NEJMoa1011923.
Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379(25):2395–406. https://doi.org/10.1056/NEJMoa1809775.
Sonbol MB, Mountjoy LJ, Firwana B, Liu AJ, Almader-Douglas D, Mody K, et al. The role of maintenance strategies in metastatic colorectal cancer: a systematic review and network meta-analysis of randomized clinical trials. JAMA Oncol. 2020;6(3):e194489. https://doi.org/10.1001/jamaoncol.2019.4489.
Acknowledgements
Not Applicable.
Funding
This on-going study is supported by an unrestricted research grant from Bayer Healthcare Pharmaceutical, which also provided regorafenib. The funder has no role in study design, management, analysis and interpretation of data as well as no role in the writing of the final report.
Author information
Authors and Affiliations
Contributions
AA, TM, CBM, and MY designed the clinical study and will contribute to data interpretation. AA, TM, LE and FG contributed to the drafting of the manuscript. PC contributed to the trial set-up. PC and SD are responsible for data collection and for administrative support. BP does the circulating cell-free DNA analyses. JF and CBM designed the statistical part of the study and will contribute to statistical analysis. All authors contributed to the revision of the manuscript and approved it for submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by French regulatory authorities (Agence nationale de sécurité du médicament et des produits de santé) on Jan 24th 2019, received a favorable opinion by the “Comité de Protection des Personnes EST III” (Dec 6th, 2018) and complies with the Helsinki declaration and French laws and regulations, and follows the International Conference on Harmonisation E6 (R2) Guideline for Good Clinical Practice. Written informed consent will be obtained from each patient before any screening and inclusion procedure. The trial results, even inconclusive, will be submitted in a peer-reviewed journal. Trial registration numbers are the following: EudraCT number: 2018–003541-42; ClinicalTrials.gov number: NCT03828799.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Adenis, A., Mazard, T., Fraisse, J. et al. FOLFIRINOX-R study design: a phase I/II trial of FOLFIRINOX plus regorafenib as first line therapy in patients with unresectable RAS-mutated metastatic colorectal cancer. BMC Cancer 21, 564 (2021). https://doi.org/10.1186/s12885-021-08312-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08312-7