
Abstract
Background
Lung adenocarcinoma (LUAD) remains a crucial factor endangering human health. Gene-based clinical predictions could be of great help for cancer intervention strategies. Here, we tried to build a gene-based survival score (SS) for LUAD via analyzing multiple transcriptional datasets.
Methods
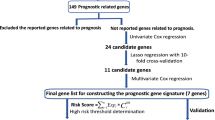
We first acquired differentially expressed genes between tumors and normal tissues from intersections of four LUAD datasets. Next, survival-related genes were preliminarily unscrambled by univariate Cox regression and further filtrated by LASSO regression. Then, we applied PCA to establish a comprehensive SS based on survival-related genes. Subsequently, we applied four independent LUAD datasets to evaluate prognostic prediction of SS. Moreover, we explored associations between SS and clinicopathological features. Furthermore, we assessed independent predictive value of SS by multivariate Cox analysis and then built prognostic models based on clinical stage and SS. Finally, we performed pathway enrichments analysis and investigated immune checkpoints expression underlying SS in four datasets.
Results
We established a 13 gene-based SS, which could precisely predict OS and PFS of LUAD. Close relations were elicited between SS and canonical malignant indictors. Furthermore, SS could serve as an independent risk factor for OS and PFS. Besides, the predictive efficacies of prognostic models were also reasonable (C-indexes: OS, 0.7; PFS, 0.7). Finally, we demonstrated enhanced cell proliferation and immune escape might account for high clinical risk of SS.
Conclusions
We built a 13 gene-based SS for prognostic prediction of LUAD, which exhibited wide applicability and could contribute to LUAD management.
Similar content being viewed by others


Background
Lung cancer remains intractable but imperative to cope with for the highest morbidity and mortality among cancers [1]. A principle subtype of lung cancers is lung adenocarcinoma (LUAD), whose investigation means a great deal to us [2,3,4]. Advance in cancer biology demonstrated cancer could be regarded as a disorder caused mainly by aberrant genes, while some core ones even drive carcinogenesis [5, 6]. That is to say, genes are undoubtedly valuable targets for cancer management.
In fact, remarkable achievements in clinical practice have proved powerful effect of genes on clinical oncology especially for LUAD [7, 8]. First take chemotherapy for example. Many widely applied chemotherapeutic agents are aimed at critical genes in biological processes like cell proliferation and metabolism [9, 10]. Besides, targeted therapy based on driver gene, such as epidermal growth factor receptor (EGFR), has significantly improved the prognosis of patients with specific genetic background [11, 12]. Moreover, immunotherapy targeted at immune-checkpoint genes has achieved revolutionary progress for LUAD patients, especially for who have no targetable driver mutation till now [4, 7, 13].
Moreover, clinical predictions based on gene signatures also contribute much to handling cancer [14,15,16]. For example, some canonical biomarkers are references for distinguishing specific cancer from normal counterparts and other histologic subtypes [17, 18]. Besides, other gene-based clinical prediction like prognostic prediction has emerged as a hot spot for the relative convenience to obtain and the great significance for treatment [15]. Tremendous advance in omics and mature application of statistical methods in bioinformation contributed greatly to bridge genes signatures and cancer characteristics. For example, The Cancer Genome Atlas (TCGA) program and Gene Expression Omnibus (GEO) database both offer abundant resources for cancer investigation. The Least Absolute Shrinkage and Selection Operator (LASSO) regression can both adjust the complexity and execute variable selection, thereby improving the prediction precision and interpretability of the regression model [19]. Moreover, compared with bio-enrichment methods based only on differentially expressed genes (DEGs), Gene Set Enrichment Analysis (GSEA) could take into account those genes with subtle expression changes but significant biological significance, therefore, it is more comprehensive and precise [20]. Herein, we tried to build a gene-based survival score (SS) for LUAD via systematic transcriptome analysis, and this SS exhibited favorable predictive efficacy in multiple datasets.
Methods
Transcriptomic and clinical information
LUAD datasets containing gene expression profiling and clinical information were obtained from TCGA program (RNA-sequencing) and GEO database (gene microarray). We applied different datasets to different analysis based on the data characteristic and analytic demands, as follows. Four datasets consisting of transcriptome profiling in tumors and normal tissues were applied for filtrating DEGs (GSE32863, 58 tumors and 58 normal tissues; GSE43458, 80 tumors and 30 normal tissues; GSE10072, 58 tumors and 49 normal tissues; TCGA-LUAD, 58 paired tumors and normal tissues) [21,22,23,24]. Four datasets containing non-controversial and available records about Overall Survival (OS) and Progression-free Survival (PFS) were applied for survival analysis (TCGA-LUAD, 402 samples; GSE30219, 85 samples; GSE31210, 200 samples; GSE50081, 124 samples) [24,25,26,27]. TCGA-LUAD was used as training set while GSE30219, GSE31210 and GSE50081 were applied as validation sets.
Besides, 515 samples of TCGA-LUAD, GSE30219, GSE31210 and GSE50081 were candidates for enrichment analysis. And 253 samples of TCGA-LUAD containing comprehensive clinicopathologic records (age, gender, TNM parameters, clinical stage, OS and PFS) were applied for multivariate analysis.
Statistical methods
DESeq2 package (RNA-sequencing) and limma package (microarrays) were applied to DEGs (Adjusted P-value < 0.05, fold change > 2 or < 0.5) [28, 29]. Z score was used to normalize data (Function: scale). Cox regression model, LASSO regression model, Kaplan-Meier (K-M) curve and log-rank test were used for survival analysis (Packages: survival, survminer and glmnet). Principal component analysis (PCA) was utilized for comprehensive assessment (Function: princomp; Packages: FactoMineR and factoextra). Receiver operating characteristic (ROC) curve analysis was performed to determine optimal cut-off (Packages: pROC). Logistic regression model was applied to find associations between genes and two-category data (TNM parameters and clinical stage were transferred to two-category data) (Function: glm). Pearson correlation analysis was applied for correlation assessment (Packages: ggcorrplot). GSEA was employed for biological investigation [20]. Wilcoxon rank sum test was applied for differential analysis between two groups (Function: wilcox.test). P < 0.05 was considered significant. Arithmetic functions were operated in R language [30].
Results
Identifying 13 core genes to establish SS for LUAD
Genes closely related to tumor prognosis are likely to play key roles in tumor progression. Valuable candidates are DEGs between tumors and normal tissues. So we obtained the intersection of DEGs from four LUAD transcriptomic datasets (GSE10072, GSE32863, GSE43458 and TCGA-LUAD), and we acquired 52 upregulated DEGs and 180 downregulated DEGs (fold change > 2 or fold change < 0.5, Adjusted P-value < 0.05) (Fig. 1a) (Detailed information about acquiring DEGs could be seen in our previous research [31]). Filtrating survival-related genes from these DEGs was executed in TCGA-LUAD dataset, for its largest sample size and most complete clinical records. We conducted univariate Cox regression analysis towards OS for preliminary identification, and we obtained 16 hazardous genes from upregulated DEGs and 35 protective genes from downregulated DEGs (for hazardous genes, which may promote cancer, p < 0.05, HR > 1; for protective genes, which may prevent cancer, p < 0.05, HR < 1) (Fig. 1b, c). Since independence among variables is a prerequisite for establishing multi-factor models, we analyzed associations among the screened genes through a correlation matrix. The results showed strong relationship among these genes, so further screening was needed (Fig. 1d). Thereupon, we implemented LASSO analysis and we got 13 core genes: Abnormal Spindle Microtubule Assembly (ASPM), Epithelial Cell Transforming 2 (ECT2), Glucosaminyl (N-Acetyl) Transferase 3, Mucin Type (GCNT3), Golgi Membrane Protein 1 (GOLM1), Insulin Like Growth Factor 2 MRNA Binding Protein 3 (IGF2BP3), Solute Carrier Family 2 Member 1 (SLC2A1), Solute Carrier Family 7 Member 5 (SLC7A5), Tissue Inhibitor Of Metalloproteinases 1 (TIMP1), Thymidylate Synthetase (TYMS), Rac/Cdc42 Guanine Nucleotide Exchange Factor 6 (ARHGEF6), Cytochrome P450 Family 4 Subfamily B Member 1 (CYP4B1), Family With Sequence Similarity 189 Member A2 (FAM189A2), and Secretoglobin Family 1A Member 1 (SCGB1A1) (Fig. 1e, f) (Coefficients of genes in LASSO regression are listed in Supplementary Table 1). Furthermore, we performed PCA for the comprehensive evaluation based on core genes (Fig. 1g). Finally, we chose the first 6 principal components (comps) to establish a SS for LUAD (The cumulative contribution rate is more than 70%):

A Survival-related SS for LUAD. a DEGs acquired from the intersection of four LUAD datasets (GSE10072, GSE32863, GSE43458 and TCGA-LUAD); b, c Survival-related genes screened out preliminarily from upregulated DEGs and downregulated DEGs according to OS via univariate Cox regression analysis; d The correlation matrix for survival-related genes screened preliminarily; e, f Core genes identified further considering OS by LASSO regression analysis; g Comprehensive assessment of core genes by PCA
Score = 0.409981*comp.1 + 0.170820*comp.2 + 0.129785*comp.3 + 0.106358*comp.4 + 0.09938*comp.5 + 0.083671*comp.6.
And coefficients for genes to make up each comp are exhibited in Supplementary Table 2.
SS exhibits high-risk probability for OS in LUAD
Next, we estimated prognostic value of SS in LUAD. We chose OS for prognostic indicator and examined in four independent LUAD datasets (TCGA-LUAD, GSE30219, GSE31210, GSE50081). We divided patients into two groups (High and Low SS) based on optimal cut-off value via ROC curve analysis according to the survival status of OS in each dataset respectively (Fig. 2a-d). We found SS showed obvious high-risk probability for OS and patients with higher SS had shorter OS periods in four datasets (Fig. 2e-h).

OS rates between LUAD patients with high and low SS. a-d ROC curves for determining optimal cut-off for SS upon OS in TCGA-LUAD (a), GSE30219 (b), GSE31210 (c) and GSE50081 (d); e-h K-M survival curves and HRs considering OS between patients with high and low SS in TCGA-LUAD (e), GSE30219 (f), GSE31210 (g) and GSE50081 (h)
SS possesses high-risk probability for PFS in LUAD
Then, we estimated predictive value of SS upon PFS in four independent LUAD datasets (TCGA-LUAD, GSE30219, GSE31210, GSE50081). We implemented ROC curve to divided patients into High and Low SS groups based on outcome status of PFS in these datasets respectively (Fig. 3a-d). Analogously, we found higher SS indicated bigger risk probability for PFS in all testing datasets (Fig. 3e-h).

PFS probabilities between LUAD patients with high and low SS. a-d ROC curves for finding appropriate cut-off for SS upon PFS in TCGA-LUAD (a), GSE30219 (b), GSE31210 (c) and GSE50081 (d); e-h K-M survival curves and HRs related to PFS between patients with high and low SS in TCGA-LUAD (e), GSE30219 (f), GSE31210 (g) and GSE50081 (h)
SS correlates highly with clinicopathological features and functions as a novel independent risk factor for LUAD prognosis
Further, we investigated relationships between SS and clinicopathological parameters of LUAD. Marker of proliferation Ki-67 (MKI67) and proliferating cell nuclear antigen (PCNA) are both canonical biomarkers for clinical oncology [32,33,34,35]. We found SS was intensively positively correlated with MKI67 in four LUAD datasets (TCGA-LUAD, GSE30219, GSE31210, GSE50081) (Fig. 4a-d). Analogously, SS possessed strong positive association with PCNA in these datasets (Fig. 4e-h). TNM parameters and tumor clinical stage also play important roles in tumor handling. And we chose TCGA-LUAD dataset with relatively more complete clinical information for following analysis. We found that SS indicated high-risk probability for N (lymph node metastasis), M (distant metastasis) and clinical stage (Fig. 4i). Usually, univariate analysis might cover up the real prognostic function due to confounding factors. So we verified further SS could function as an independent risk predictor for both OS and PFS via multiple Cox regression analysis considering age, gender, TNM parameters and clinical stage in TCGA-LUAD (Table 1, 2). Moreover, we used clinical stage and SS to build concise nomographs predicting OS and PFS probability of LUAD (Fig. 4j, k). And predictive potencies were acceptable (C-indexes: OS, 0.7; PFS, 0.7).

Relationships between SS and clinicopathological features of LUAD. a-d Correlations between SS and MKI67 expression in TCGA-LUAD (a), GSE30219 (b), GSE31210 (c) and GSE50081 (d); e-h Correlations between SS and PCNA expression in TCGA-LUAD (e), GSE30219 (f), GSE31210 (g) and GSE50081 (h); i ORs regarding TNM parameters and clinical stage between patients with high and low SS; j The nomograph for predicting one-year and five-year OS probability; k The nomograph for predicting one-year and five-year PFS probability
Exploring molecular characteristics underlying SS in LUAD
We tried to uncover molecular mechanisms underlying clinical role of SS in LUAD. We first ranked the patients (515 samples in TCGA-LUAD) in order of SS. The top 50 patients were divided into High SS group, and last 50 patients were Low SS group (Fig. 5a). Then we performed GSEA to investigate molecular features of SS based on transcription profiling. We found high SS showed enhanced cell cycle in several gene sets (Fig. 5b). Further, we validated it in other three datasets (GSE30219, top 40 vs last 40; GSE31210, top 50 vs last 50; GSE50081, top 50 vs last 50), and found similar results (Fig. 5c-h). Last, we explored expression profile of immune checkpoints under SS in above four LUAD datasets, and found most of immune checkpoints possessed increased expression in high SS group (Fig. 5i-l).

Molecular characteristics underlying SS in LUAD. a, c, e, g Dividing LUAD patients into high and low group based on SS in TCGA-LUAD (a), GSE30219 (c), GSE31210 (e) and GSE50081 (g); b, d, f, h Gene sets enriched in high SS group from several collections of the MSigDB (Only top ten significant gene sets were presented) in TCGA-LUAD (b), GSE30219 (d), GSE31210 (f) and GSE50081 (h); i-l Immune-check genes expression between patients with high and low SS in TCGA-LUAD (i), GSE30219 (j), GSE31210 (k) and GSE50081 (l)
Discussion
LUAD possesses strong heterogeneities in both tumor biology and clinical characteristics [36]. Therefore, it is urgently needed to precisely assess LUAD prognosis for applying appropriate intervention as well as avoiding overtreatment. Deregulation of gene expression during malignant transformation and progression offers theoretical basis to interpret carcinogenesis via gene signatures, while the tremendous advance in onco-genomics provides prominently practical convenience [6, 14, 16]. Here, we established a gene-based survival assessment named SS, which exhibited good accuracy and stability in multiple datasets.
Compared with former gene-based prognostic predictions [37,38,39,40], SS owns three specialties or innovations: 1. SS has favorable stability, or adaptability in clinical application. Our initial candidates are common DEGs between tumors and normal tissues in four independent LUAD datasets. After screening by Cox model and LASSO regression, we established a 13 gene-based SS in TCGA-LUAD, and validated its efficiency in other three LUAD datasets. 2. SS was multifunction for clinical usage. First, SS could assess both OS and PFS. OS is a golden standard of prognosis evaluation but takes a long time to collect, while PFS is a relatively convenient indicator for clinical intervention. Besides, SS was positively related to malignant biomarkers and tumor stage, and could function as an independent risk factor for prognosis. 3. Biological significance of SS was verified in multiple datasets. In four LUAD datasets, higher SS all indicated enhanced cell proliferation, which confirmed the prime trait of abnormal proliferation in carcinogenesis. Moreover, LUAD with higher SS exhibited increased expression of immune checkpoint genes, which underlined the prominent role of immune escape in malignant progression of LUAD.
These 13 core genes building SS are involved in a diversity of biological processes like cell proliferation, nutrition transportation and material metabolism. Most of these genes have been reported to play critical roles in lung carcinogenesis, however, some lack detailed research. For example, ASPM and ECT2 both are proliferation-related genes and participate in DNA synthesis and cytokinesis [41,42,43]. Studies have shown that LUAD had high expression of ASPM and ECT2, which both indicated poor prognosis, and ECT2 could facilitate lung tumorigenesis [43,44,45,46,47,48,49]. Lung cancer also has high expression profile of GCNT3 (a gene regulating mucin synthesis), GOLM1(a gene coding for a Golgi transmembrane protein) and IGF2BP3 (a gene coding for a RNA binding protein), which are all positively correlated with malignant progression of lung cancer [50,51,52,53]. Nutrient transporter SLC2A1 and SLC7A5 both exhibit cancer-promoting potential in lung cancer [54,55,56]. TIMP1 was originally thought to be a tumor suppressor gene, since it could intensely inhibit matrix metalloproteinases (MMPs), canonical oncoproteins [57, 58]. However, recent studies have shown that TIMP1 was highly expressed in LUAD, functioned as an independent prognostic risk factor and could facilitate malignant progression through non-MMPs pathways [58,59,60]. TYMS, a nucleotide synthetase, is commonly used as an indicator of chemotherapy sensitivity, that is its high expression in lung cancer often indicates insensitive for pemetrexed [61]. Also, for patients under platinum-based adjuvant treatment after surgical resection, high TYMS expression often indicates poorer prognosis [62]. SCGB1A1, coding for secretory globulins, has a protective role against smoking-induced lung tumorigenesis [63]. However, for CYP4B1, ARHGEF6 and FAM189A2, their function in lung cancer are rarely studied.
Of course, there are some flaws in our research. First, the present transcriptomic data mainly covered protein-coding genes, but increasing studies have uncovered that non-coding RNAs like microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and circulating RNAs (cirRNAs) present powerful biological functions, especially in cancer research [64]. Second, the transcriptomic data for analysis came from tissues, while estimation based on liquid detection would be less invasive and more executable [65]. Improvement will be made in our future work.
Conclusions
In brief, we built a gene-based survival SS for LUAD and proved its wide applicability for clinical predictions, which will assist in handling LUAD effectively.
Availability of data and materials
Original data can be found in https://www.ncbi.nlm.nih.gov/geo/ and https://portal.gdc.cancer.gov/.
Abbreviations
- LUAD:
-
Lung adenocarcinoma
- SS:
-
Survival score
- EGFR:
-
Epidermal growth factor receptor
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- DEGs:
-
Differentially expressed genes
- GSEA:
-
Gene Set Enrichment Analysis
- OS:
-
Overall Survival
- PFS:
-
Progression-free Survival
- K-M:
-
Kaplan-Meier
- PCA:
-
Principal component analysis
- ROC:
-
Receiver operating characteristic
- ASPM:
-
Abnormal Spindle Microtubule Assembly
- ECT2:
-
Epithelial Cell Transforming 2
- GCNT3:
-
Glucosaminyl (N-acetyl) Transferase 3, Mucin Type
- GOLM1:
-
Golgi Membrane Protein 1
- IGF2BP3:
-
Insulin Like Growth Factor 2 MRNA Binding Protein 3
- SLC2A1:
-
Solute Carrier Family 2 Member 1
- SLC7A5:
-
Solute Carrier Family 7 Member 5
- TIMP1:
-
Tissue Inhibitor Of Metalloproteinases 1
- TYMS:
-
Thymidylate Synthetase
- ARHGEF6:
-
Rac/Cdc42 Guanine Nucleotide Exchange Factor 6
- CYP4B1:
-
Cytochrome P450 Family 4 Subfamily B Member 1
- FAM189A2:
-
Family With Sequence Similarity 189 Member A2
- SCGB1A1:
-
Secretoglobin Family 1A Member 1
- comps:
-
Principal components
- MKI67:
-
Marker of proliferation Ki-67
- PCNA:
-
Proliferating cell nuclear antigen
- miRNAs:
-
MicroRNAs
- lncRNAs:
-
Long non-coding RNAs
- cirRNAs:
-
Circulating RNAs
- AUC:
-
Area under the curve
- CI:
-
Confidence interval
- MSigDB:
-
Molecular Signatures Database
- NES:
-
Normalized Enrichment Score
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Thomas A, Liu SV, Subramaniam DS, Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol. 2015;12(9):511–26.
Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, Shepherd FA. Non-small-cell lung cancer. Lancet (London, England). 2011;378(9804):1727–40.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–54.
Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell. 2018;173(2):371–85 e318.
Roychowdhury S, Chinnaiyan AM. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J Clin. 2016;66(1):75–88.
Chalela R, Curull V, Enríquez C, Pijuan L, Bellosillo B, Gea J. Lung adenocarcinoma: from molecular basis to genome-guided therapy and immunotherapy. J Thorac Dis. 2017;9(7):2142–58.
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50.
Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Rev Clin Oncol. 2014;11(5):282–98.
Bailly C. Irinotecan: 25 years of cancer treatment. Pharmacol Res. 2019;148:104398.
Reck M, Rabe KF. Precision diagnosis and treatment for advanced non-small-cell lung Cancer. N Engl J Med. 2017;377(9):849–61.
Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr, Wu YL, Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet (London, England). 2017;389(10066):299–311.
Saito M, Suzuki H, Kono K, Takenoshita S, Kohno T. Treatment of lung adenocarcinoma by molecular-targeted therapy and immunotherapy. Surg Today. 2018;48(1):1–8.
Cieslik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet. 2018;19(2):93–109.
Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173(2):400–16 e411.
Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nat Med. 2011;17(3):297–303.
Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018;173(2):291–304 e296.
Sung HJ, Cho JY. Biomarkers for the lung cancer diagnosis and their advances in proteomics. BMB Rep. 2008;41(9):615–25.
Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J Royal Stat Soc Series B-Statistical Methodol. 2011;73:273–82.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22(7):1197–211.
Kabbout M, Garcia MM, Fujimoto J, Liu DD, Woods D, Chow CW, Mendoza G, Momin AA, James BP, Solis L, et al. ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin Cancer Res. 2013;19(13):3383–95.
Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3(2):e1651.
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer genome atlas pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–20.
Rousseaux S, Debernardi A, Jacquiau B, Vitte AL, Vesin A, Nagy-Mignotte H, Moro-Sibilot D, Brichon PY, Lantuejoul S, Hainaut P, et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5(186):186ra66.
Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72(1):100–11.
Der SD, Sykes J, Pintilie M, Zhu CQ, Strumpf D, Liu N, Jurisica I, Shepherd FA, Tsao MS. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J Thorac Oncol. 2014;9(1):59–64.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Chan BKC. Data analysis using R programming. Adv Exp Med Biol. 2018;1082:47–122.
Xiong Y, Lei J, Zhao J, Feng Y, Qiao T, Zhou Y, Jiang T, Han Y. Gene expression-based clinical predictions in lung adenocarcinoma. Aging. 2020;12(15):15492–503.
Martin B, Paesmans M, Mascaux C, Berghmans T, Lothaire P, Meert AP, Lafitte JJ, Sculier JP. Ki-67 expression and patients survival in lung cancer: systematic review of the literature with meta-analysis. Br J Cancer. 2004;91(12):2018–25.
Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182(3):311–22.
Stoimenov I, Helleday T. PCNA on the crossroad of cancer. Biochem Soc Trans. 2009;37(Pt 3):605–13.
Lv Q, Zhang J, Yi Y, Huang Y, Wang Y, Wang Y, Zhang W. Proliferating cell nuclear antigen has an association with prognosis and risks factors of Cancer patients: a systematic review. Mol Neurobiol. 2016;53(9):6209–17.
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–46.
Zhao K, Li Z, Tian H. Twenty-gene-based prognostic model predicts lung adenocarcinoma survival. OncoTargets and therapy. 2018;11:3415–24.
Wistuba II, Behrens C, Lombardi F, Wagner S, Fujimoto J, Raso MG, Spaggiari L, Galetta D, Riley R, Hughes E, et al. Validation of a proliferation-based expression signature as prognostic marker in early stage lung adenocarcinoma. Clin Cancer Res. 2013;19(22):6261–71.
Robles AI, Arai E, Mathe EA, Okayama H, Schetter AJ, Brown D, Petersen D, Bowman ED, Noro R, Welsh JA, et al. An integrated prognostic classifier for stage I lung adenocarcinoma based on mRNA, microRNA, and DNA methylation biomarkers. J Thorac Oncol. 2015;10(7):1037–48.
Song Q, Shang J, Yang Z, Zhang L, Zhang C, Chen J, Wu X. Identification of an immune signature predicting prognosis risk of patients in lung adenocarcinoma. J Transl Med. 2019;17(1):70.
Capecchi MR, Pozner A. ASPM regulates symmetric stem cell division by tuning Cyclin E ubiquitination. Nat Commun. 2015;6:8763.
Higgins J, Midgley C, Bergh AM, Bell SM, Askham JM, Roberts E, Binns RK, Sharif SM, Bennett C, Glover DM, et al. Human ASPM participates in spindle organisation, spindle orientation and cytokinesis. BMC Cell Biol. 2010;11:85.
Bai X, Yi M, Xia X, Yu S, Zheng X, Wu K. Progression and prognostic value of ECT2 in non-small-cell lung cancer and its correlation with PCNA. Cancer Manag Res. 2018;10:4039–50.
Li L, Peng M, Xue W, Fan Z, Wang T, Lian J, Zhai Y, Lian W, Qin D, Zhao J. Integrated analysis of dysregulated long non-coding RNAs/microRNAs/mRNAs in metastasis of lung adenocarcinoma. J Transl Med. 2018;16(1):372.
Zhang Y, Wang H, Wang J, Bao L, Wang L, Huo J, Wang X. Global analysis of chromosome 1 genes among patients with lung adenocarcinoma, squamous carcinoma, large-cell carcinoma, small-cell carcinoma, or non-cancer. Cancer Metastasis Rev. 2015;34(2):249–64.
Bidkhori G, Narimani Z, Hosseini Ashtiani S, Moeini A, Nowzari-Dalini A, Masoudi-Nejad A. Reconstruction of an integrated genome-scale co-expression network reveals key modules involved in lung adenocarcinoma. PLoS One. 2013;8(7):e67552.
Sun Y, Wang L, Jiang M, Huang J, Liu Z, Wolfl S. Secreted phosphoprotein 1 upstream invasive network construction and analysis of lung adenocarcinoma compared with human normal adjacent tissues by integrative biocomputation. Cell Biochem Biophys. 2010;56(2–3):59–71.
Zhou S, Wang P, Su X, Chen J, Chen H, Yang H, Fang A, Xie L, Yao Y, Yang J. High ECT2 expression is an independent prognostic factor for poor overall survival and recurrence-free survival in non-small cell lung adenocarcinoma. PLoS One. 2017;12(10):e0187356.
Justilien V, Ali SA, Jamieson L, Yin N, Cox AD, Der CJ, Murray NR, Fields AP. Ect2-dependent rRNA synthesis is required for KRAS-TRP53-driven lung adenocarcinoma. Cancer Cell. 2017;31(2):256–69.
Li Q, Ran P, Zhang X, Guo X, Yuan Y, Dong T, Zhu B, Zheng S, Xiao C. Downregulation of N-Acetylglucosaminyltransferase GCNT3 by miR-302b-3p decreases non-small cell lung Cancer (NSCLC) cell proliferation, migration and invasion. Cell Physiol Biochem. 2018;50(3):987–1004.
Liu X, Chen L, Zhang T. Increased GOLM1 expression independently predicts unfavorable overall survival and recurrence-free survival in lung adenocarcinoma. Cancer Control. 2018;25(1):1073274818778001.
Li W, Li N, Gao L, You C. Integrated analysis of the roles and prognostic value of RNA binding proteins in lung adenocarcinoma. PeerJ. 2020;8:e8509.
Yan J, Wei Q, Jian W, Qiu B, Wen J, Liu J, Fu B, Zhou X, Zhao T. IMP3 predicts invasion and prognosis in human lung adenocarcinoma. Lung. 2016;194(1):137–46.
Guo W, Sun S, Guo L, Song P, Xue X, Zhang H, Zhang G, Li R, Gao Y, Qiu B, et al. Elevated SLC2A1 expression correlates with poor prognosis in patients with surgically resected lung adenocarcinoma: a study based on Immunohistochemical analysis and bioinformatics. DNA Cell Biol. 2020;39(4):631–44.
Zhang B, Xie Z, Li B. The clinicopathologic impacts and prognostic significance of GLUT1 expression in patients with lung cancer: a meta-analysis. Gene. 2019;689:76–83.
Takeuchi K, Ogata S, Nakanishi K, Ozeki Y, Hiroi S, Tominaga S, Aida S, Matsuo H, Sakata T, Kawai T. LAT1 expression in non-small-cell lung carcinomas: analyses by semiquantitative reverse transcription-PCR (237 cases) and immunohistochemistry (295 cases). Lung cancer (Amsterdam, Netherlands). 2010;68(1):58–65.
Ramer R, Bublitz K, Freimuth N, Merkord J, Rohde H, Haustein M, Borchert P, Schmuhl E, Linnebacher M, Hinz B. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. FASEB J. 2012;26(4):1535–48.
Jackson HW, Defamie V, Waterhouse P, Khokha R. TIMPs: versatile extracellular regulators in cancer. Nat Rev Cancer. 2017;17(1):38–53.
Selvaraj G, Kaliamurthi S, Lin S, Gu K, Wei DQ. Prognostic impact of tissue inhibitor of Metalloproteinase-1 in non- small cell lung Cancer: systematic review and meta-analysis. Curr Med Chem. 2019;26(42):7694–713.
Xiao W, Wang L, Howard J, Kolhe R, Rojiani AM, Rojiani MV. TIMP-1-Mediated Chemoresistance via Induction of IL-6 in NSCLC. Cancers. 2019;11(8):1184.
Chamizo C, Zazo S, Dómine M, Cristóbal I, García-Foncillas J, Rojo F, Madoz-Gúrpide J. Thymidylate synthase expression as a predictive biomarker of pemetrexed sensitivity in advanced non-small cell lung cancer. BMC pulmonary medicine. 2015;15:132.
Sun S, Shi W, Wu Z, Zhang G, Yang BO, Jiao S. Prognostic significance of the mRNA expression of ERCC1, RRM1, TUBB3 and TYMS genes in patients with non-small cell lung cancer. Experimental and therapeutic medicine. 2015;10(3):937–41.
Yang Y, Zhang Z, Mukherjee AB, Linnoila RI. Increased susceptibility of mice lacking Clara cell 10-kDa protein to lung tumorigenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a potent carcinogen in cigarette smoke. J Biol Chem. 2004;279(28):29336–40.
Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. 2019;179(5):1033–55.
Bardelli A, Pantel K. Liquid biopsies, what we do not know (yet). Cancer Cell. 2017;31(2):172–9.
Acknowledgments
Not applicable.
Funding
The National Natural Science Foundation of China (81772462) provided funding support to the design, analysis and publication of this research.
Author information
Authors and Affiliations
Contributions
YLX conceived the study, performed analysis and wrote the original draft. JL and JBZ conducted data curation, language correction and revised the manuscript. QL, YBF, TYQ and SWX helped validation and formal analysis. YH and TJ supervised research, acquired financial support and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Because the data were obtained from TCGA and GEO database, all ethics approvals and informed consents to participate were already obtained by the original studies.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table 1
, Coefficients for genes under LASSO regression.
Additional file 2: Supplementary Table 2
, Coefficients for genes to make up each comp.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiong, Y., Lei, J., Zhao, J. et al. A gene-based survival score for lung adenocarcinoma by multiple transcriptional datasets analysis. BMC Cancer 20, 1046 (2020). https://doi.org/10.1186/s12885-020-07473-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-07473-1