
Abstract
Background
Esophageal squamous cell carcinoma (ESCC) is an aggressive cancer with one of the highest world incidences in the Eastern Cape region of South Africa. Several genome wide studies have been performed on ESCC cohorts from Asian countries, North America, Malawi and other parts of the world but none have been conducted on ESCC tumors from South Africa to date, where the molecular pathology and etiology of this disease remains unclear. We report here tumor associated copy number changes observed in 51 ESCC patients’ samples from the Eastern Cape province of South Africa.
Methods
We extracted tumor DNA from 51 archived ESCC specimens and interrogated tumor associated DNA copy number changes using Affymetrix® 500 K SNP array technology. The Genomic Identification of Significant Targets in Cancer (GISTIC 2.0) algorithm was applied to identify significant focal regions of gains and losses. Gains of the top recurrent cancer genes were validated by fluorescence in situ hybridization and their protein expression assessed by immunohistochemistry.
Results
Twenty-three significant focal gains were identified across samples. Gains involving the CCND1, MYC, EGFR and JAG1 loci recapitulated those described in studies on Asian and Malawian cohorts. The two most significant gains involved the chromosomal sub-bands 3q28, encompassing the TPRG1 gene and 11q13.3 including the CTTN, PPFIA1and SHANK2 genes. There was no significant homozygous loss and the most recurrent hemizygous deletion involved the B3GAT1 gene on chromosome 11q25. Focal gains on 11q13.3 in 37% of cases (19/51), consistently involved CTTN and SHANK2 genes. Twelve of these cases (23,5%), had a broader region of gain that also included the CCND1, FGF19, FGF4 and FGF3 genes. SHANK2 and CTTN are co-amplified in several cancers, these proteins interact functionally together and are involved in cell motility. Immunohistochemistry confirmed both Shank2 (79%) and cortactin (69%) protein overexpression in samples with gains of these genes. In contrast, cyclin D1 (65%) was moderately expressed in samples with CCND1 DNA gain.
Conclusions
This study reports copy number changes in a South African ESCC cohort and highlights similarities and differences with cohorts from Asia and Malawi. Our results strongly suggest a role for CTTN and SHANK2 in the pathogenesis of ESCC in South Africa.
Similar content being viewed by others

Background
Esophageal squamous cell carcinoma (ESCC) is an aggressive cancer which occurs in specific regions of the world which include Lixian China, Japan, the Golestan province of Iran, parts of South America (Uruguay) and the eastern corridor of Africa, (Malawi, Kenya and South Africa (SA) [1,2,3]. In South Africa, the Eastern Cape province has one of the highest world incidences of 31.3 and 18 per 100,000 male and female individuals respectively [4]. A number of early studies in western countries have identified ESCC risk factors such as alcohol consumption and smoking. However, these risk factors are absent in a number of high endemic areas where other causes, including nutritional deficiencies, lower socio-economic status, consumption of hot beverages and exposure to polycyclic aromatic hydrocarbons are suspected [2, 3]. ESCC risk has also been related to the consumption of maize contaminated by aflatoxin [5, 6] and in South Africa, chronic inflammation caused by a local cultural practice of induced vomiting, was thought to play a role [7]. The respective impact of these factors is unclear and environmental/cultural exposures are likely to interact with population specific genetic susceptibilities. The dismal prognosis of this disease [third cause of death in SA [8], and first cause of death in both males and females in the Eastern Cape region (unpublished data from community-based cancer registry)] underscores the need to understand its molecular pathology.
Several genome-wide copy number studies have been performed on ESCC cohorts from Asian and western countries, using technologies of varied resolutions. The most recurrent somatic copy number variations (SCNV) across these studies involve gains on chromosomes 3q26-q29, 7p11.2-p22.1, 8q22.3–24.21, 11q12.3-q13.4 and 20q11-q13.33 and losses on chromosomes 3p11.1–14.2, 8p21.3-p23.2, 9p21.3–24.1 and 18q11-q22.3. These regions host key cancer genes including PIK3CA, SOX2, EGFR, MYC, CCND1, CTTN, FHIT and CDKN2A/B [9,10,11,12,13,14]. The most common recurrent gains across studies involves the 11q12.3–13.4 region with amplicons of varied size that almost always include the oncogene CCND1 [9,10,11,12,13,14,15]. Apart from copy number aberrations, mutational analyses have shown recurrent inactivating mutations in TP53, and NOTCH1 as well as activating events in PIK3CA [10, 11, 15]. A single genomic study, performed on African patients from Malawi, recapitulated patterns of gene mutations and copy number changes (gains of CCND1, TP63, MYC, ERBB2, EGFR, MYCL1 and losses of CDKN2A/CDKN2B), similar to those observed in Asian and North American ESCC patients [16]. Of note, gene expression patterns from transcriptome sequence analysis in this African cohort highlighted three distinct ESCC subgroups that tended to reflect exposure to differing environmental factors [16]. The diversity in the genomic landscape observed in this study strongly warrants the expansion of genomic investigations in other African countries with high ESCC incidence in order to infer etiologic factors and identify markers of disease with a potential for early detection and improved therapeutic interventions.
Apart from a report using conventional cytogenetic comparative genomic hybridization (CGH) [17], and a study on five ESCC cell-lines established in SA [18], there are no high-resolution genome wide SCNV data on ESCC in South Africa. We report SCNVs in 51 ESCC tumor specimens derived from a single geographic region of South Africa that shows one of the highest world incidences for this disease.
Methods
Tumor material and patient characteristics
Eighty-two archived, formalin fixed paraffin embedded (FFPE) ESCC specimens were collected from the archives of the Nelson Mandela Academic Hospital in Mthatha, Eastern Cape from the years 2004–2006. The ratio of males to females was 1:1.16. Haematoxylin and eosin stained slides were reviewed and marked by an experienced pathologist to identify tumor areas (> 80% tumor cells) for DNA extraction. Thirty FFPE samples with a normal tissue histology from a matched population (age and ethnicity) were collected from the same laboratory and constituted the reference panel for copy number analysis.
Genomic DNA isolation
Tumors and control specimens were pre-treated in 1 M sodium thiocyanate and DNA was extracted using proteinase K digestion followed by phenol/chloroform extraction. DNA quality was assessed by standard gel electrophoresis and spectrophotometry. FFPE DNA is known to show varying degrees of degradation and to establish the ability of these samples to amplify large fragments, a multiplex PCR assay (previously described) was performed prior to array processing [19]. Of 82 ESCC samples collected, 51 yielded enough quality DNA to proceed with SNP arrays.
Affymetrix 500 K SNP array
DNA from ESCC and control specimens were hybridized to Affymetrix® 250 K Nsp and Sty GeneChips® respectively, which have a mean probe spacing of 5.8 kb. Samples were hybridized once per chip type. The Affymetrix® GeneChip® mapping 500 K protocol (P/N 701930 Rev. 3) was followed, apart from the number of PCR reactions per sample, which was increased to six to yield the optimal amount of 90 μg of PCR product. Scanning was performed on the Affymetrix® GeneChip Scanner 3000 7G (Affymetrix®, Santa Clara USA). The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus [20] and are accessible through GEO Series accession number GSE59105 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59105).
500 K data analysis
Raw intensity data (CEL files) were imported into Genotyping Console™ (Affymetrix®, Santa Clara USA) to assess the SNP call rates as an initial quality control measure. The average call rates were 71.3 and 72.1% for Nsp and Sty respectively. Call rates were expected to be lower than for fresh tissue (93–95%) due to poor amplification of larger fragments during PCR [21]. The raw intensity data of 50 samples were imported into Partek® Genomics Suite where quantile normalization, SNPs on fragments larger than 700 bp were removed and copy number analysis were performed. The copy number data were segmented using the circular binary algorithm in GenePattern [22] using a minimum of 10 markers for regions of gain and loss. Common copy number variants were removed from the data after comparing each region of change to the Database of Genomic Variants (http://projects.tcga.ca/variation). To assess the significance of gains and losses, the segmentation file was analysed using GISTIC 2.0 ref (Genomic Identification of Significant Targets in Cancer) [23] using a q-value cut-off of 0.25.
Common regions of gain or loss and the respective genes involved were reported using the Refseq database, genome build hg18.
Fluorescence in situ hybridisation (FISH)
Gains of CCND1, and MYC were validated on 10 samples using the LSI t(11;14) dual color dual translocation probe (Abbott Molecular, USA), which covers the CCND1 and FGF4 loci on chromosome 11 and the LSI MYC SpectrumOrange probe (Abbott Molecular, USA) respectively. BAC clones were obtained from the BACPAC resource center, Children’s Hospital Oakland Research Institute, CA, USA. The BAC clone, RP11-736 L3 (Chr 11: 70,732,999-70,899,011), mapping to SHANK2 gene on 11q13.3 was labeled by nick translation with SpectrumOrange-dUTP (Abbott Molecular, USA) and hybridized to 10 ESCC samples as described previously [18]. Briefly, three-micron sections were baked at 60 °C overnight and de-waxed twice in Xylene (Merck). Dehydrated slides were pre-treated in 0.2 N HCl for 20 min, followed by 1 M sodium thiocyanate at 80 °C for 30 min. Air dried slides were treated with Pepsin (Roche) (0.5 mg/ml) for 20 min to 1 h30 minutes at 37 °C depending on the tissue size and thickness. Slides were rinsed in 2x SSC, dried at 42 °C and fixed in 1% formaldehyde at room temperature. Pre-treated samples were denatured in 50% formamide buffer at 76 °C for 5 min, dehydrated in ice-cold ethanol and denatured probes (76 °C for 5 min) were added for overnight hybridisation at 37 °C. The next day, slides were washed in 2x SSC at 76 °C for 5 min, counterstained with DAPI and mounted using Vectashield® fluorescent mounting medium (Vectalabs, USA). Images were captured using Cytovision 4.0 (Applied Imaging) on an Olympus BX61 fluorescent microscope.
Immunohistochemistry (IHC)
In order to assess the protein expression of the most recurrent target genes, we performed immunohistochemistry on 4 μm deparaffinised sections in duplicate. The DAKO EnVision FLEX detection system was used according to the manufacturer’s instructions. Cyclin D1 was detected using ready-to use FLEX monoclonal anti-cyclin D1 (Clone EP12, Dako IR08361) as supplied. The Cortactin and Shank2 proteins were detected using rabbit monoclonal anti-cortactin antibody (EP1922Y, Abcam, 0.095 mg/ml) diluted to 1:250 and rabbit polyclonal anti-Shank2 antibody (aa 331–380, Abcam, 1 mg/ml) diluted to 1:75 respectively. Slides were counterstained with Haematoxylin and mounted in aqueous mounting solution. Positive controls were respectively, breast tumour for Cortactin, mantle cell lymphoma for Cyclin D1 and staining observed in suprabasal epithelial cells of normal oesophageal squamous epithelium for Shank2. The primary antibody was replaced with antibody diluent as a negative control. To correlate the gains of SHANK2, CCND1 and CTTN genes with their respective protein expression, samples with gains of these 3 genes (n = 22), gains of SHANK2 alone (n = 2) and no gains (n = 2) were processed. Staining was scored on the intensity (0–3) and the percentage of positive cells (0 = no staining, 1 = < 10% with moderate staining, 2= > 10% with moderate staining and 3 ≥ 50% with intense staining.
Results
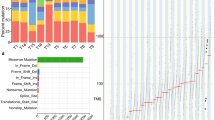
Array copy number analysis of South African ESCC samples revealed a high level of complexity in the tumor genome with most chromosomes showing aberrations, (median number of aberrations per case: 96, minimum: 33, maximum: 426). GISTIC 2.0 analysis identified 30 gains (Supplementary Table 1) and 36 deletions (Supplementary Table 2) (Fig. 1a and b).

Summary of gains and loss identified by GISTIC 2.0. a Copy number gains identified in ESCC by GISTIC 2.0. b Copy number deletions detected by GISTIC 2.0. c Graph representing focal gains (≤3 genes) identified by GISTIC 2.0 analysis sorted by frequency. d Graph showing focal deletions (≤3 genes) detected by GISTIC 2.0 analysis sorted by frequency
Gains
Twenty-three focal gains (≤3 genes) were observed (Table 1). Those involved chromosomes 1q31.1, 1p31.3, 2p24.2, 2q24.3, 3q28, 4q13.3, 5p13.2, 6p24.3, 7p11.2, 8p12, 8p23.2, 8q24.12, 8q24.21, 9p21.1, 10p11.21, 11q13.3, 12q14.1, 13q22.1, 14q23.2, 15q11.2, 19q12, 20p12.2 and 20q13.2. The two top recurrent gains involved
the TPRG1 gene on 3q28 (21/51 cases, 41%), and the CTTN, PPFIA1 and SHANK2 genes on 11q13.3 (19/51, 37%) (Fig. 1c). Although the function of the TPRG1 gene is not well established, amplification and/or activating mutations in Cis regulatory elements of this gene associated with its increased expression have recently been reported in diffuse large B-cell lymphomas, suggesting potential oncogenic activity [24].
Chromosome 11q13.3 gain is a common event in ESCC, where it almost always involves the CCND1 proto-oncogene [9,10,11, 13] and, to a lesser extent, the CTTN and SHANK2 genes. In our cohort CTTN and SHANK2 were the most frequent amplified genes at 11q13.3 and this region expanded proximally to include the CCND1, FGF19, FGF4 and FGF3 in 12 / 51 cases.
The cortactin protein, encoded by the CTTN gene, is an actin binding scaffolding protein with various cellular functions and is known to promote cell motility [25]. The Shank2 protein belongs to another family of scaffolding proteins and is a cortactin binding partner [26]. It has mostly been studied in neuronal synapses and its role in cancer is unclear [27]. Similarly, the PPFIA1 gene, which encodes the cytosolic scaffolding protein lyprin-α1 [28], is a potential target gene often co-amplified at 11q13.3 with CCND1 and the above two genes in ESCC [29].
CCND1 encodes a protein which promotes cell cycle progression. Gain thereof and associated increased expression is well described in a variety of cancer types including head and neck squamous cell carcinoma and ESCC [13,14,15,16, 30].
Other notable significant focal gains involved the known proto-oncogenes EGFR and MYC on 7p11.2 and 8q24.21 respectively (Table 1). EGFR copy gains are seen in approximately 20% of ESCC patients, who show improved survival when treated with the anti-EGFR kinase inhibitor, gefitinib [31].
FISH confirmed gains of SHANK2 and CCND1 in 10 cases and matched closely with array analysis data (Fig. 2).

CCND1/FGF4 and SHANK2 genes copy number. (A) DAPI stained nuclei from sample UROC171. a1 FISH analysis was performed with the Vysis LSI t(11;14) dual color probe. The IGH gene probe on chromosome 14, acts as an internal control (green signal), the red signal represents locus specific probe encompassing the CCND1 and FGF4 genes). Gains are seen with 6–8 red signals (white arrow) while the control probe shows two green signals in most cells. a2 DAPI stained nuclei from UROC171 case, hybridized with the BAC clone, RP11-736 L3 (SHANK2 gene), labeled with SpectrumOrange-dUTP (Abbott Molecular, USA). Clumping of red signals for SHANK2 (white arrow), were consistent with high-level gains. This type of signal pattern was approximated to 20 signals. b 500 K SNP copy number segmentation for chromosome 11q in all samples generated in GenePattern (IGV). The minimal common region of gain (11q13.3: 69889604–70,002,885) is represented by the red box. This region includes the CTTN, PPFIA1 and SHANK2 genes. c Graphs showing the average copy number of CCND1 and SHANK2 for each of the 10 samples analyzed by FISH. c1 The average CCND1 copy number across 10 samples was 15.7 by FISH and 16,5 by copy number array analysis (11q: 68884395-70,061,246 bp) in the same cases. c2 Gain of SHANK2 was confirmed by FISH in 10 cases (average of 14,2 copies), the same cases had an average copy number of 23,5 by array copy number (11q:70,061,246-70,310,057)
Evaluation of cyclin D1, Shank2 and cortactin proteins expression
To assess if the most common gains resulted in increased protein expression of target genes, we assessed Shank2 and cortactin immunoreactivity in normal and tumor esophageal tissues. Signals for both proteins were low in non-neoplastic esophageal squamous epithelium, in the cytoplasm (Shank2) or nuclei (cortactin), of basal epithelial cells, and disappeared in cells leaning towards the luminal surface (Fig. 3). Twenty-six tumor samples were assessed for Shank2, cortactin and cyclin D1 protein expression; of these, 22 cases had DNA gain of all three genes and 19/22 (86%) overexpressed Shank2 (score3), 16/22 (72%) overexpressed cortactin, while only 5/22 cases (22%) overexpressed cyclin D1, (score of 3). Cyclin D1 was moderately expressed in 12/22 cases (54%) (score of 2) (Fig. 3, panel a). Overall, 19/26 (73%) and 18/26 (69%) of cases overexpressed Shank2 and cortactin respectively. One case had gain of CCND1 only, but all three genes showed moderate protein expression on IHC. One sample with SHANK2 gain only, overexpressed Shank2 as well as cortactin, while cyclin D1 was moderately expressed (Fig. 3, Panel b). One case had no gains of these three genes and over expressed cortactin, while Shank2 and cyclin D1 were weakly expressed (score 1). In summary, Shank2 and cortactin were co-expressed in most cases with gains of these genes. Co-amplification of CTTN, SHANK2 and CCND1 genes has been reported previously in oral squamous cell carcinoma. In contrast to our study all cases overexpressed cyclin D1 (quantitative PCR analysis), while a subset of cases 50% overexpressed CTTN and SHANK2 [32].

Representative images of the common immunohistochemical staining patterns for Shank2, cortactin and cyclin D1. a shows Shank2 staining (40x magnification) in non-neoplastic oesophageal squamous mucosa, cytoplasmic signal was observed in basal cells, which disappeared towards the luminal surface. b shows staining of CCND1 in non-neoplastic oesophageal squamous mucosa (40x magnification), staining was observed in nuclei, which disappeared towards the luminal surface. Panel A: Case UROC48 with co-amplification of the SHANK2, CTTN and CCND1 genes. a) shows intense cytoplasmic staining for Shank2 (score 3). b) intense cytoplasmic and membranous staining for cortactin (score 3). c) Moderate staining for cyclin D1 (score 1). Panel B: Case UROC144 with amplification of the SHANK2 gene only. a) shows intense cytoplasmic staining for Shank2 (score 3), b) shows intense cytoplasmic staining for cortactin (score 3) and c) shows moderate staining for cyclin D1 (score 2)
Losses
Twelve significant focal deletions were detected by GISTIC 2.0 analysis (Table 2 and Fig. 1d). All losses were heterozygous. These deletions covered chromosomal regions 1p36.32, 2p21, 4q35.1, 5q33.2, 8q24.3, 10p15.3, 11q25, 12p13.33, 13q34, 14q23.3, 15q13.1 and 22q13.33. The most frequent losses were on chromosome 11q25 (67%) and 10p15.3 (66%). Both regions covered one gene, B3GAT1 and ADARB2 respectively. ADARB2 has no known role in cancer. B3GAT1, also known as CD57, expression was previously tested in 3672 prostate cancer and benign specimens by IHC. While CD57 was expressed in benign prostate and low-grade prostate cancer, loss of expression correlated with tumor de-differentiation and size [33]. Three other regions of loss harbored genes with a known tumor suppressor function. These included the ZFP36L2 gene on 2p21, ING2 on 4q23.3 as well as the microRNA MIR625, and FUT8 gene on 14q23.3. ZFP36L2 is a putative transcription factor involved in cellular responses, which was shown to act as a tumor suppressor in colorectal cancer and acute myeloid leukemia [34, 35]. Lack of expression of the known tumor suppressor ING2, a chromatin remodeling protein, has been reported in several types of cancer [reviewed in [36]]. Decreased expression of MIR625 was described in colorectal carcinoma. Expression of this microRNA in colorectal metastatic models in nude mice was shown to suppress cell invasion and metastasis suggesting a tumor suppressor activity [37]. Decreased expression of MIR625 was reported in ESCC patients previously where it was associated with a 5-year decreased survival rate (38.1%) compared to ESCC patients with higher MIR625 expression [38].
Discussion
We determined the pattern of segmental gains and losses in ESCC tumors from South African patients of the Eastern Cape Province, a region with one of the highest ESCC incidences in the world, using high resolution 500 K SNP array technology. Our results showed both differences and similarities in SCNVs compared to studies performed on ESCC cohorts form Asia and Malawi. The high number (96 mean aberrations per case) and heterogeneous nature of SCNVs was in keeping with the notion that ESCC is a genetically complex disease [9,10,11, 13].
Large-scale gains on chromosomes 3q, 8q and 11q, observed in this study were similar to those reported previously [9,10,11,12,13,14]. One of the most frequent (88%) common focal regions of high copy gain on chromosome 11q13 observed here almost always involved the CTTN, SHANK2 and PPFIA1genes.
The SHANK2 and CTTN genes are in close proximity (30 kb) and are often co-amplified in oral squamous cell carcinoma [32]. These two genes’ protein products interact together and in its epithelial isoform, Shank2 binds to the SH3 domain of cortactin. Shank2-cortactin interaction was shown to facilitate cell motility by preventing anoikis through the PI3-Akt pathway in neural cells [27, 39]. One can hypothesise that such interaction may occur in ESCC thus facilitating cell motility and metastasis. CTTN gain/ increased expression alone has been associated with ESCC metastasis and functional studies further demonstrated that inhibition of CTTN expression decreased tumor growth and lung metastasis [27]. Additionally, two previous studies reported overexpression of CTTN in ESCC pre-cancerous lesions [40, 41]. In addition, in the 11q13.3 region of focal gain, the PPFIA1 gene has not been studied extensively in ESCC but was shown to be significantly overexpressed in head and neck squamous cell carcinoma [42].
In our South African cohort, 12/51 cases had a broader region of gain on chromosome 11q13.3, which included the known oncogenes CCND1, FGF3, FGF4, FGF19 as well as the recently described oncogenic MIR548K [10]. This broader region of gain has been described in a number of previous investigations including in 5 ESCC cell-lines established in South Africa [9,10,11,12,13,14,15,16,17,18]. In our cohort, cyclin D1 expression correlated to a lesser extent with gains of CCND1 (5/23 cases) than Shank2 and cortactin. CCND1 remains an important candidate in ESCC as a known oncogene involved in a number of malignancies and as a notable cell cycle regulator [13, 42]. MIR548K, shown to enhance cell proliferation in ESCC cell-lines [13], may also be a candidate key gene considering that this micro RNA lies within the broader region of gain on chromosome 11q13 in the present cohort.
The significant region of focal gain detected on chromosome 3q28, targeted the TPRG1 (tumor protein p63 regulated 1) gene. Although this gene has not been linked to ESCC pathogenesis, its distal neighbor gene, TP63 showed gains in a wider peak region, in 20 of the 21 cases with gains at 3q28. TP63 is a significant target of 3q gain in ESCC patients from Malawi as well as in ESCC cohorts from Western and Asian countries [16, 43]. Of note, TPRG1 is highly expressed in normal esophageal tissue and an intergenic susceptibility locus (rs6791479) was identified in a genome-wide association study of cutaneous squamous cell carcinoma in between the TP63 and TPRG1 genes [44]. Taken together with the fact that the ESCC genomic profile is closer to other squamous cell carcinomas than to esophageal adenocarcinoma, the above observations support the notion that one or both these genes may play an important role in South African ESCC pathogenesis [43].
Chromosome 3q amplicons have been described across a number of ESCC studies and usually involve the PIK3CA and/or SOX2 genes [9, 10, 12, 14]. By contrast to the cohort in Malawi, these genes did not show copy number alteration in our cases [16]. Mutational analysis would have to be performed to exclude activating mutations.
Significant gains involving the oncogene MYC were observed in our cohort, in keeping with studies that implicated the 8q24.1-q24.2 chromosomal region in other populations [9, 10, 13, 14, 16]. Similarly, gains involving the EGFR gene at chromosome 7p11.2 are previously described and thought to play a role in ESCC pathophysiology [9, 10, 13, 16, 18].
There were no significant homozygous deletions in this series as per GISTIC 2.0 analysis. Of note, no losses at the CDKN2A, CDKN2B and TP53 loci were detected in this cohort in contrast with losses observed in the cohort from Malawi [16]. Although this could be due to incorrect array normalization, it is unlikely since our FISH results correlated tightly with arrays results.
We acknowledge limitations of this study due to the lack of patients’ clinical data and that aberrations detected could not be correlated with risk factors endemic to the region. No correlation could be established between copy number variants and stages of disease. Genome wide mutational analysis was also not performed in the present study and is currently being conducted on South African samples as part of a larger international collaboration.
Conclusions
This study describes both common and differing regions of copy number aberrations in ESCC from South Africa when compared to other cohorts. Of note, our results suggest a role for Shank2 and cortactin proteins in ESCC carcinogenesis in South Africa. This will have to be clarified by future functional studies with a view to developing new markers of disease.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in NCBI’s Gene Expression Omnibus [20] and are accessible through GEO Series accession number GSE59105 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59105).
Abbreviations
- BAC:
-
Bacterial artificial chromosome
- ESCC:
-
Esophageal squamous carcinoma
- FFPE:
-
Formalin fixed paraffin embedded
- FISH:
-
Fluorescence in situ hybridisation
- GEO:
-
Gene Expression Omnibus
- GISTIC:
-
Genomic Identification of Significant Targets in Cancer
- IHC:
-
Immunohistochemistry
- PCR:
-
Polymerase chain reaction
- SCNV:
-
Somatic cop number variants
- SNP:
-
Single nucleotide polymorphism
- SSC:
-
Saline sodium citrate
References
Islami F, Kamangar F, Nasrollahzadeh D, Møller H, Boffetta P, Malekzadeh R. Oesophageal cancer in Golestan Province, a high-incidence area in northern Iran - a review. Eur J Cancer. 2009;45(18):3156–65.
Abnet CC, Arnold M, Wei W. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154(2):360–73.
Murphy G, McCormack V, Abedi-Ardekani B, et al. International cancer seminars: a focus on esophageal squamous cell carcinoma. Ann Oncol. 2017;28:2086–93.
Somdyala NI, Bradshaw D, Gelderblom WCA, Parkin DM. Cancer incidence in a rural population of South Africa, 1998-2002. Int J Cancer. 2010;127(10):2420–9.
Pacella-Norman R, Urban MI, Sitas F, Carrara H, Sur R, Hale M, Ruff P, Patel M, Newton R, Bull D, Beral V. Risk factors for oesophageal, lung, oral and laryngeal cancers in black south Africans. Br J Cancer. 2002;86(11):1751–6.
Sun G, Wang S, Hu X, Su J, Huang T, Yu J, Tang L, Gao W, Wang J. Fumonisin B1 contamination of home-grown corn in high-risk areas for esophageal and liver cancer in China. Food Addit Contam. 2007;24(2):181–5.
Matsha T, Stepien A, Blanco-Blanco E, Brink LT, Lombard CJ, van Rensburg S, Erasmus RT. Self-induced vomiting – risk for oesophageal cancer? S Afr Med J. 2006;96(3):209–2012.
GLOBOCAN 2012: Estimated cancer incidence mortality and prevalence worldwide in 2012. http://globocan.iarc.fr/Pages/fact_sheets_population.aspx.
Hu N, Wang C, Ng D, Clifford R, Yang HH, Tang Z, Wang Q, Han X, Giffen C, Goldstein AM, Taylor PR, Lee MP. Genomic characterization of esophageal squamous cell carcinoma from a high-risk population in China. Cancer Res. 2009;69(14):5908–17.
Song Y, Li L, Ou Y, Gao Z, Li E, Li X, Zhang W, Wang J, Xu L, Zhou Y, Ma X, Liu L, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509(91):92–6.
Lin D, Hao J, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding L, Garg M, Liu L, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46(5):467–75.
Bandla S, Pennathur A, Luketich JD, Beer DG, Lin L, Bass A, Godfrey TE, Litle VR. Comparative genomics of esophageal adenocarcinoma and squamous cell carcinoma. Ann Thorac Surg. 2012;93(4):1101–6.
Ying J, Shan L, Li J, Zhong L, Xue L, Zhao H, Li L, Langford C, Guo L, Qiu T, Lu N, Tao Q. Genome-wide screening for genetic alterations in esophageal cancer by aCGH identifies 11q13 amplification oncogenes associated with nodal metastasis. PLoS One. 2012;7(6):e39797.
Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, et al. SOX2 is an amplified lineage survival oncogene in lung and esophageal squamous carcinomas. Nat Genet. 2009;41(11):1238–42.
Dai W, Ko JMY, Choi SSA, Yu Z, Ning L, Zheng H, Gopalan V, Chan KT, Lee NP, Chan KW, Law SY, Lam AK, Lung ML. Whole-exome sequencing reveals critical genes underlying metastasis in oesophageal squamous cell carcinoma. J Pathol. 2017;242:500–10.
Liu W, Snell JM, Jeck WR. Subtyping sub-Saharan esophageal squamous cell carcinoma by comprehensive molecular analysis. JCI Insight. 2016;1(16):e88755.
Du Plessis L, Dietzsch E, Van Gele M, Van Roy N, Van Helden P, Parker IM, Mugwanya DK, De Groot M, Marx MP, Kotze MJ, Speleman F. Mapping of novel regions of DNA gain and loss by comparative genomic hybridization in esophageal carcinoma in the black and colored populations of South Africa. Cancer Res. 1999;59(8):1877–83.
Brown J, Bothma H, Veale R, Willem P. Genomic imbalances in esophageal carcinoma cell lines involve Wnt pathway genes. World J Gastroenterol. 2011;17(24):2909–23.
van Beers EH, Joosse SA, Ligtenberg MJ, Fles R, Hogervorst FBL, Verhoef S, Nederl PF. A multiplex PCR predictor for aCGH success of FFPE samples. Br J Cancer. 2006;94(2):333–7.
Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;1(3):207–10.
Jacobs S, Thompson ER, Nannya Y, Yamamoto G, Pillai R, Ogawa S, Bailey DK, Campbell IG. Genome-wide, high resolution detection of copy number, loss of heterozygosity and genotypes from formalin-fixed, paraffin embedded tumour tissue using microarrays. Cancer Res. 2007;67(6).
Reich M, Gould J, Lerner J, Tamayo P, Mesirov JP. Genepattern 2.0. Nat Genet. 2006;38(5):500–1.
Mermel CH, Shumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC 2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41.
Cornish A, Hoang J, Dobbins PH, Law SE, Chubb PJ, Orlando D, Houlston G. Identification of recurrent noncoding mutations in B-cell lymphoma using capture hi-C. Blood Adv. 2019;3(1):21–32.
Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773(5):642–52.
Weaver AM. Cortactin and tumor invasiveness. Cancer Lett. 2008;265(2):157–66.
MacGillavry HD, Kerr JM, Kassner J, Frost NA, Blanpied TA. Shank–cortactin interactions control actin dynamics to maintain flexibility of neuronal spines and synapses. Eur J Neurosci. 2016;43(2):179–93.
Pehkonen H, von Nandelstadh P, Karhemo PR, et al. Liprin- alpha1 is a regulator of vimentin intermediate filament network in the cancer cell adhesion machinery. Sci Rep. 2016;6:24486.
Chang J, Tan W, Ling Z, Xi R, et al. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat Commun. 2017;8:15290.
Ramos-García P, González-Moles MÁ, Ayén Á, González-Ruiz L, Gil-Montoya JA, Ruiz-Ávila I. Predictive value of CCND1/cyclin D1 alterations in the malignant transformation of potentially malignant head and neck disorders: systematic review and meta-analysis. Head Neck. 2019;41(9):3395–407.
Petty RD, Dahle-Smith A, Stevenson DAJ, Osborne A, Massie D, Clark C, Murray GI, Dutton SJ, Roberts C, Chong IY, Mansoor W, Thompson J, Harrison M, Chatterjee A, Falk SJ, Elyan S, Garcia-Alonso A, Fyfe DW, Wadsley J, Chau I, Ferry DR, Miedzybrodzka Z. Gefitinib and EGFR gene copy number aberrations in esophageal Cancer. J Clin Oncol. 2017;35(20):2279–87.
Freier K, Sticht C, Hofele C, Flechtenmacher C, Stange D, Puccio L, Toedt G, Radlwimmer B, Lichter P, Freier SJ. Recurrent Coamplification of cytoskeleton-associated genes EMS1 and SHANK2 with CCND1 in Oral squamous cell carcinoma. Genes Chromosom Cancer. 2006;45:118–25.
Wangarin H, Kristiansen G, Schlomm T, Stephan C, Gunia S, Zimpfer A, Weichart W, Sauter G, Erbersdobler A. CD57 Expression in Incidental, Clinically Manifest, and Metastatic Carcinoma of the Prostate. BioMed Res Int. 2014:356427.
Suk F, Chang C, Lin R, et al. ZFP36L1 and ZFP36L2 inhibit cell proliferation in a cyclin D-dependent and p53-independent manner. Nat Sci Rep. 2018;8:2742.
Liu J, Lu W, Liu S, et al. ZFP36L2, a novel AML1 target gene, induces AML cells apoptosis and inhibits cell proliferation. Leuk Res. 2018;68:15–21.
Ludwig S, Klitzsch A, Baniahmad A. The ING tumor suppressors in cellular senescence and chromatin. Cell Biosci. 2011;1:25.
Lou X, Qi X, Zhang Y, Long H, Yang A. Decreased expression of microRNA-625 is associated with tumor metastasis and poor prognosis in patients with colorectal Cancer. J Surg Oncol. 2013;108:230–5.
Li C, Li D, Che S, Ma K, Wang Y, Xia L, Dai X, Zhang G, Shen Y, Jiao W, Tian K. The decreased expression of miR-625 predicts poor prognosis of esophageal squamous cell carcinoma. Int J Clin Exp Med. 2015;8(6):9560–4.
Du Y, Weed SA, Xiong WC, Marshall TD, Parsons JT. Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol Cell Biol. 1998;18(10):5838–51.
Hsu KF, Lin CK, Yu CP, Tzao C, Lee SC, Lee YY, Tsai WC, Jin JS. Cortactin, fascin, and survivin expression associated with clinicopathological parameters in esophageal squamous cell carcinoma. Dis Esophagus. 2009;22(5):402–8.
Hsu NY, Yeh KT, Chiang IP, Pai LY, Chen CY, Ho HC. Cortactin overexpression in the esophageal squamous cell carcinoma and its involvement in the carcinogenesis. Dis Esophagus. 2008;21(5):402–8.
Zhou H, Cao T, Li WP, Wu G. Combined expression and prognostic significance of PPFIA1 and ALG3 in head and neck squamous cell carcinoma. Mol Biol Rep. 2019;46(3):2693–701.
Network CGAR, Analysis Working Group: Asan University, BC Cancer Agency; Brigham and Women’s Hospital, et al. Integrated genomic characterization of oesophageal carcinoma. Nature. 2017;541(7636):169–75.
Asgari MM, Wang W, Ioannidis NM, Itnyre J, Hoffmann T, Jorgenson E, Whittemore AS. Identification of susceptibility loci for cutaneous squamous cell carcinoma. J Invest Dermatol. 2016 May;136(5):930–7.
Acknowledgements
We would like to thank Antony Holmes for re-mapping the segmentation file to match marker files for GISTIC 2.0 analysis and Penny Keene for critically reviewing this manuscript.
Funding
Funding was provided by the Cancer Association of South Africa (CANSA), the Medical Research Council of South Africa (MRC) and the National Health Laboratory Services. These funding bodies provided financial support only and did not contribute to the study design, analysis, interpretation or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
JB performed all the experimental procedures, analyzed the data and wrote the manuscript. AS collected the specimens and reviewed the histopathology of all cases. PW conceptualized the study, coordinated the study, contributed to analysis and wrote the manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study received ethics approval from the University of the Witwatersrand human research ethics committee, in accordance with the Declaration of Helsinki (Reference number: M090658). These samples were retrospective FFPE samples obtained from the archive of the histopathology department. Samples could not be linked to living individuals and therefore consent could not be obtained. The samples were de-identified for the purpose of this study to preserve patient anonymity in accordance with the regulations of the local ethics committee.
Consent for publication
Consent for publication was waived as this study was a retrospective study performed on archived tissue samples that could not be linked to living individuals.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Supplementary Table 1. Table of all gains detected by GISTIC 2.0. Supplementary Table 2. Table of deletions detected by GISTIC 2.0
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Brown, J., Stepien, A.J. & Willem, P. Landscape of copy number aberrations in esophageal squamous cell carcinoma from a high endemic region of South Africa. BMC Cancer 20, 281 (2020). https://doi.org/10.1186/s12885-020-06788-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-06788-3