
Abstract
Background
Invasive urothelial carcinoma (iUC) is highly similar between dogs and humans in terms of pathologic presentation, molecular subtypes, response to treatment and age at onset. Thus, the dog is an established and relevant model for testing and development of targeted drugs benefiting both canine and human patients. We sought to identify gene expression patterns associated with two primary types of canine iUC tumors: those that express a common somatic mutation in the BRAF gene, and those that do not.
Methods
We performed RNAseq on tumor and normal tissues from pet dogs. Analysis of differential expression and clustering, and positional and individual expression was used to develop gene set enrichment profiles distinguishing iUC tumors with and without BRAFV595E mutations, as well as genomic regions harboring excessive numbers of dysregulated genes.
Results
We identified two expression clusters that are defined by the presence/absence of a BRAFV595E (BRAFV600E in humans) somatic mutation. BRAFV595E tumors shared significantly more dysregulated genes than BRAF wild-type tumors, and vice versa, with 398 genes differentiating the two clusters. Key genes fall into clades of limited function: tissue development, cell cycle regulation, immune response, and membrane transport. The genomic site with highest number of dysregulated genes overall lies in a locus corresponding to human chromosome 8q24, a region frequently amplified in human urothelial cancers.
Conclusions
These data identify critical sets of genes that are differently regulated in association with an activating mutation in the MAPK/ERK pathway in canine iUC tumors. The experiments also highlight the value of the canine system in identifying expression patterns associated with a common, shared cancer.
Similar content being viewed by others
Background
Urothelial carcinoma is the second most common cancer of the urinary tract in humans following prostate cancer [1]. Approximately 80,000 new cases are diagnosed each year with 17,670 people expected to die from the disease this year [2]. The invasive form of urothelial carcinoma (iUC), which comprises 25–30% of human bladder cancer cases, is the most common urinary bladder tumor of dogs. It accounts for ≥90% of canine bladder tumors, with ≥50,000 dogs predicted to develop the disease yearly in the U.S. alone [3, 4]. In dogs, the tumors are naturally occurring, with the majority being high-grade papillary infiltrative tumors [3]. Distant metastases are present in about 15–20% of dogs at diagnosis, and in ≥50% of dogs at death, with liver, lung, and bone being frequent sites of metastases [3,4,5,6].
Invasive urothelial carcinoma is similar between dogs and humans in terms of pathologic presentation including cellular features and tumor heterogeneity, molecular subtypes (basal and luminal), and response to treatment [1, 4, 7]. The most common clinical signs at presentation in dogs include blood in the urine, frequent and painful urination and frequent small voids [7]. Diagnosis of iUC is made by histologic assessment of biopsies obtained via cystoscopy or surgery. While complete cystectomy is often a front-line treatment for human iUC, it is rarely used in dogs as tumor growth into the urethra is common at presentation, as is metastasis. The morbidity and cost associated with the procedure is a further deterrent for both surgeons and patient families. Instead, the most common treatment includes COX inhibitors and chemotherapy, offered together or separately [7, 8].
In the past several years canine iUC has been established as a relevant model for the testing and development of targeted drugs that benefit both canine and human patients [3, 9, 10].. Because of similarities in molecular features, tumor heterogeneity, metastatic behavior, and an immunocompetent host, the dog system closely mimics the human condition. It is, thus, expected that the canine system will predict drug outcomes in humans with high fidelity. Examples of targeted agents currently undergoing testing in dogs include folate-vinblastine, [11] folate-tubulysin, [12] and 5-azacitadine [13].
Genomic analyses of human iUC reveal multiple tumor subtypes, identified through expression profiles, often combined with somatic mutations and/or genomic rearrangements [14,15,16,17,18,19,20,21]. As few as two, and as many as nine, overlapping clusters of human iUC tumors have been identified, with results highly dependent on study design factors such as analysis methods, number of tumors analyzed and types of data collected [14,15,16,17,18,19,20,21]. Despite variation across studies, there is strong evidence that clustering patterns can predict disease progression and treatment response [14, 22,23,24]. This is due to the occurrence of some common clusters across studies including the division between luminal and basal-like tumors and the identification of expression-based clusters related to immune function that indicate infiltration of the tumors by immunologic cells including lymphocytes, macrophages, and myofibroblasts [16, 17, 20].
Two studies of gene expression in canine iUC, one based on expression arrays and another using RNAseq identified a similar basal/luminal division as that observed in human iUC cancers [25, 26]. Two additional canine iUC RNAseq studies, however, which included seven and 11 tumors, respectively, reported no distinct clusters, although both reported similarities between canine and human tumor expression profiles, including altered expression in genes or pathways involved in cell cycle progression, DNA repair, and inflammation [27, 28]. These differences indicate that there is much we still don’t understand about iUC specific expression profiles in dogs, and additional consideration is needed which uses a genome-wide, rigorous and unbiased approach to delineating regulation of genes critical to expression profiles, as we have done here.
Herein we have sequenced the transcriptomes of 15 canine iUC tumors and identified two expression clusters that can be defined by the presence or absence of a BRAFV595E somatic mutation which we have previously observed in > 85% of canine iUC tumors [29, 30]. The same mutation in humans (termed BRAFV600E) is present in several human cancer types including melanoma, colorectal cancer, thyroid cancer, and hairy-cell leukemia [31]. In this study we present the primary sources of differentiation between the two tumor clusters, both in terms of types of genes altered as well as regions of the genome harboring excessive numbers of dysregulated genes.
Methods
Sample collection
Sample collection was performed following approval of the Animal Care and Use Committees of the collecting institutions, and owners of all participating dogs signed an informed consent document. Five tumor samples were obtained through cystoscopy as directed by evaluation and treatment protocols decided upon following owner/oncologist consultation. The remaining tumor samples and adjacent normal tissues were collected during necropsy when the owners elected to have their dogs euthanized due to disease progression. Appropriate care was taken to collect tissues within 30 min of time-of-death. Tissues were histopathologically confirmed to be iUC. Upon collection, tissues were snap frozen and stored at − 80 °C until processed for analysis. Whole blood samples were collected in 3–6 ml EDTA or ACD tubes and stored at 4 °C prior to DNA extraction. Genomic DNA was extracted using established protocols [32]. DNA and RNA were extracted from flash frozen tissue samples using the AllPrep kit (Qiagen Corp., Alameda, CA). All samples were stripped of identifiers, numerically coded, and aliquoted for long-term storage at − 80 °C. Tumors were genotyped for the previously described BRAFV595E somatic mutation using amplicon resequencing and/or restriction fragment length polymorphism (RFLP) digestion as previously described [29].
RNAseq
RNA samples from tumors and adjacent normal tissue samples were selected that exceeded a Bioanalyzer (Agilent, Santa Clara, CA) quality score of seven and quantified using a Qubit fluorometer (Qiagen, Alameda, CA). Unstranded RNA-Seq libraries were constructed from 1 μg total RNA using the Illumina TruSeq RNA Sample Prep Kit, v2 (Illumina, Inc., San Diego, CA). The resulting cDNA was fragmented using a Covaris E210 (Covaris, Inc., Woburn, MA). Library amplification was performed using 10 cycles to minimize the risk of over-amplification. Unique single-index barcode adapters were applied to each library. Libraries were pooled in equimolar ratio and sequenced together on an Illumina HiSeq 2500 with v4 flow cells and sequencing reagents (San Diego, CA). At least 37 million 127-base read pairs were generated for each individual library. Data was processed using RTA 1.18.61 and CASAVA 1.8.2 (Illumina, Inc., San Diego, CA). The RNAseq data is available in the NCBI short-read archive (SRA) under BioProject ID PRJNA559406.
Sequence reads were aligned to the CanFam3.1 reference using the STAR 2-part aligner [33], with genome resources generated from the CanFam3.1 fasta file downloaded from UCSC genome browser [34]. The initial guided alignment was performed using an Ensembl GTF reference file while documenting all splice junctions observed. The genome was regenerated including all known splice junctions and any novel junctions that occurred in a minimum of five reads. A second alignment was performed using the regenerated genome.
BRAFV595E mutation status was rechecked using base calls-per-read at the genomic position chr16:8296284. One matched normal sample that displayed mutant alleles at this position and one tumor that conflicted with original genotype results were excluded from further analysis. Because some matched normal tissues were used as controls, we verified the presence of distinct non-overlapping normal and tumor clusters using principal component analysis (PCA) and multidimensional scaling (MDS) of genome wide expression values before proceeding with differential expression analysis. The final sample set consisted of 15 tumors and five normal tissue samples of which three were from unaffected adjacent tissue and two were normal urothelial tissue. To perform a replication analysis we selected a comparable dataset of 12 tumors, eight with and four without BRAFV595E mutations, and four normal tissue samples available from previously published data [26]. Samples were not combined for a single analysis due to differences in RNAseq methodologies.
Differential expression and clustering
HTSeq [35] was used to perform counts of all reads aligned to each of 32,704 genes annotated in Ensembl CanFam3.1.95, and for 13,173 predicted non-coding transcripts [36]. Genes that were not expressed were removed from the analysis leaving a final total of 32,938 predicted genes (24,509 Ensembl genes + 8429 predicted non-coding genes without Ensembl IDs). Both variance stabilizing and regularized log transformation was used to normalize counts for clustering by PCA and MDS. Clustering was performed both with and without normal samples and on the Ensembl and predicted non-coding genes, independently. ConsensusCluster was used to determine cluster membership based on stability of assignment after sub-sampling the data 1000 times [37].
Differential expression between tumors and normal tissues was calculated using DESeq2 [38]. A gene was considered significantly over or under expressed if the absolute value of the log2fold change was greater-than or equal-to one and the False Discovery Rate (FDR) adjusted p-value (q-value) was < 0.01. We chose a slightly higher than usual baseline for significance (p = .01 rather than .05) to increase confidence given small numbers of samples.
Positional expression
Each gene was assigned a genomic position equal to the average of the positions centered between the first and last base in all annotated transcripts, as per Ensembl CanFam3.1.95 or CanFam3.1plus [36] The genome was divided into one Mb siding windows, overlapping by 750 Kb and gene counts within each segment were based on the assigned position. Using a hypergeometric distribution we calculated a p-value for over-representation of up- or down-regulated genes in each Mb window using the R script phyper. This was repeated for upregulated and down regulated genes, independently, in each dataset. For overlapping windows with significant p-values, the distances were summed, and p-values calculated for the entire region.
Individual expression
To determine the significance of changes in expression level of each gene per individual, z-scores were calculated for each gene by comparison to the mean and standard deviation of the normal tissue expression level after variance stabilizing transformation, similar to that described previously [39]. A z-score of +/− 2.5 was required to indicate a significant change in expression. Transcripts per million (TPM) were also calculated per gene for each sample by correcting the read counts for the average coding length of the gene in each individual sequence [40].
Gene-set enrichment and regulatory predictions
Approved symbols of genes that appeared dysregulated by at least two-fold, with a corrected p-value of less than 0.01, were compared to compiled gene lists indicated below to identify over-representation of any collated gene group in the tumor expression data. The systems used were the GSEA MutSig database version 6.2 [41, 42] which uses hypergeometric distribution to assess enrichment of genes from collated gene sets within a list of dysregulated genes, and Ingenuity Pathway Analysis (IPA) [43], which uses both the list of genes and their relative expression values to predict activity of upstream regulatory genes. The top-ten results by q-value are reported for the hallmark and curated gene sets (MutSig) and top-ten bias corrected z-scores with p-values <.05 for the upstream regulators (IPA).
Results
Sequencing coverage and QC
The complete transcriptomes of seven histologically confirmed canine InvTCC tumors carrying the BRAFV595E mutation (BRAFV595E), four tumors lacking the mutation (BRAFwt), and three matched normal tissues were sequenced to an average of 45.9 million reads per sample (range 39.4–60.7 million). These data were combined with the transcriptomes of four tumors and two normal tissue samples that were previously published [29], bringing the total number of samples to 15 tumors and five normal tissue samples. Clinical characteristics of this tumor set is described in Table 1.
Differential expression
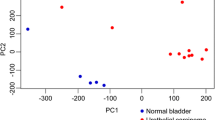
Coverage was calculated per gene for a total of 45,877 genes, which includes both protein coding and noncoding genes such as long noncoding RNAs and antisense RNAs. Of those, 32,938 were expressed in the tumor and/or normal urothelial tissues. Comparing data from tumors to that from normal samples, 3587 genes annotated in Ensemble v1.95 were differentially expressed (DE), as were 778 predicted, non-coding genes (Additional Files 1 and 2). PCA and MDS clustering of both expression datasets separated normal versus tumor samples along the first dimension and tumors carrying the BRAFV595E mutation from the BRAFwt tumors along the second dimension (Fig. 1a). When normal samples were removed from the analysis, the tumors split into two distinct clusters comprised of those with the BRAFV595E mutation and those without (Fig. 1b). Consensus clustering assigned the tumors to each cluster with an item consensus score averaging > 0.99 (range 0.965 to 1.0) (Fig. 1c).

Canine iUC tumors cluster into two distinct groups. a) Principal component analysis divides normal from tumor tissues. b) After removing the normal tissues, the first principal component separates tumors with BRAFV595E mutations from those that do not carry the mutation. c) Consensus clustering based on expression data shows two clusters of tumors. Horizontal line under dendrogram: light blue = BRAFV595E tumors, dark blue = BRAFwt tumors. Heatmap blue shades from light to dark with increased confidence. d) Distribution of Jaccard similarity scores among BRAFV595E tumors (coral), BRAFwt tumors (light blue) and between the two mutation types (green)
To confirm that expression signatures differed significantly between the BRAFV595E and BRAFwt tumors, gene expression was scored relative to normal tissues for each tumor sample independently. After assembling an individual list of genes for each sample with significant z-scores, Jaccard similarities were calculated for all tumor pairs. BRAFV595E tumors shared significantly more DE genes with each other than with BRAFwt tumors (pvalue = 7.4e-13). Similarly, BRAFwt tumors share significantly more DE genes with each other than with BRAFV595E tumors (pvalue = 0.0033) (Fig. 1d). This pattern is also observed using up- or down-regulated genes separately (Additional File 3: Supplementary Fig. 1).
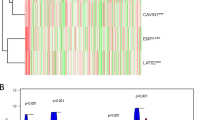
Following determination of significantly different expression patterns in BRAFV595E and BRAFwt tumors, differential expression analysis was performed for each group independently, comparing to normal tissue samples. The BRAFV595E cluster of tumors displayed 3839 DE genes, with a minimum two-fold increase or 0.5 reduction in expression (log2fold ≥1 or ≤ − 1) and Benjamini–Hochberg corrected p-values of < 0.01; 1576 up- and 2263 down-regulated. Using the same metrics, BRAFwt tumors were differentially expressed at 3724 genes; 1727 up- and 1997 down-regulated. Comparing the two tumor clusters, 2025 genes were similarly expressed in both, and only eight genes show significant expression in the opposite direction between the clusters. There are 398 genes that are significantly over- or under-expressed in one cluster with the reverse or unchanged expression (− 0.5 < log2fold change < 0.5) in the other. This minimal set of genes differentiates the two tumor clusters (Fig. 2a, Additional File 4).

Expression profile of 398 genes separates the BRAFV595E tumors from BRAFwt tumors. a) Heatmap of canine iUC tumor expression from 398 genes that are oppositely expressed between the two tumor clusters. Tumor samples are clustered by Euclidean distance, genes are clustered by correlation. Expression levels are normalized compared to expression from normal tissues. b) Top 15 GO terms over-represented in the 398 genes
Overrepresentation of gene groups
The 398 genes form three clades with similar expression patterns within each tumor cluster. These clades largely comprise genes with four primary functions; tissue development and cell cycle/cell death (64/122 genes in gene clade 1), immune response (24/85 genes in gene clade 2) and plasma membrane and membrane transport (43/104 genes in gene clade 3). These functions are also the top three identified in the enrichment analysis of the 398 genes (Fig. 2b). The immune response gene clade is upregulated in BRAFwt tumors. Tissue development genes are upregulated in BRAFV595E tumors and down regulated in BRAFwt tumors. Cell cycle and cell death are upregulated in the BRAFV595E tumors, and the membrane associated genes are down-regulated (Fig. 2a). The genes from these functional groups comprise 42% (131) of the canine iUC tumor cluster identifying genes with unique human orthologs (Additional File 5).
To confirm our findings, we assessed the expression of the 131 genes from these top functional groupings in a dataset of RNAseq results from eight BRAFV595E tumors and four BRAFwt tumors published previously [26]. We were able to divide these data into two hierarchical clusters based on Euclidian distance, with eight of eight BRAFV595E tumors in one cluster and three of four BRAFwt tumors in a second cluster. The division of genotypes among the two clusters is significantly different than random (Fishers exact pvalue = 0.0182)(Additional File 3: Supplementary Fig. 2).
Positional expression patterns
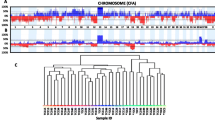
Assessing the expression of genes based on chromosomal position reveals 25 non-overlapping regions on 15 chromosomes. Each region is significantly over-represented by either up- or down-regulated genes in one or both canine iUC tumor clusters (Fig. 3a &b, Table 2). Two chromosomal regions show a lack of expression in both BRAFV595E and BRAFwt tumors, and five show increased expression in both tumor types. Ten loci are unique to the BRAFwt tumors (eight up- and two down-regulated), six are unique to the BRAFV595E tumors (five up- and one down-regulated). Thirteen of the 25 regions are syntenic to regions of common copy number variants in human tumors [44].

Positional analysis identifies loci on 15 chromosomes that contain significant numbers of dysregulated genes. The graphs are divided by a) upregulated genes and b) down regulated genes. The x-axis indicates the chromosomes from 1 to X. The y-axis showss the p-value for over-representation of dysregulated genes within a 1 Mb region surrounding each point on the graph. The dotted line indicates the Bonferroni corrected p-value of 0.05. BRAFwt tumors are shown in orange, BRAFV595E tumors are shown in blue. c) The core genes from the up-regulated locus on chr38, syntenic to the human 1q23.3 copy number variant, are overexpressed in canine iUC tumors that do not carry the BRAFV595E mutation. Boxplots display the transcripts per million compared to normal tissues from BRAFwt (orange) and BRAFV595E (blue) tumors
The single locus with the highest number of dysregulated genes is on canine chromosome 13 between 37 and 38 Mb, with 33 out of 83 genes upregulated in the BRAFwt tumors and 21 out of 83 upregulated in BRAFV595E tumors. This pattern of upregulation extends over 2.5 Mb region in BRAFwt tumors and encompasses a 1.5 Mb region in BRAFV595E tumors, yielding a total of 45 and 28 genes with increased expression, respectively (pvalue = 8.4e− 22 and 2.9e− 10). This region corresponds to the human chromosome 8q24.3, a locus commonly amplified in multiple human tumor types, including urothelial cancer [44,45,46].
One of the loci upregulated specifically in BRAFwt tumors spans 20.50 to 22.24 Mb on canine chr38, a region syntenic to human chromosome 1q23.3. The region contains 21 upregulated genes. Chromosome 1q23.3 is amplified in > 50% of human iUCs and has been associated with increased tumor stage and grade [47] as well as decreased survival time [48]. Of the nine genes that comprise the core of the human amplification region [44], seven are upregulated in BRAFwt tumors compared to normal tissue and display significantly higher expression than BRAFV595E tumors (Fig. 3c and Additional File 3: Supplementary Fig. 3). Nectin4, which is commonly used as a marker for amplification of the human 1q23.3 locus demonstrates the highest levels of expression in the BRAFwt tumors compared to normal tissue samples (log2fold = 5.89, adjP = 4.09e− 10). Nectin-4 protein is a cell surface adhesion molecule previously named poliovirus receptor-related 4 (PVRL4). The canine protein is 94% identical to the human, which is highly expressed in 60% of human bladder tumors [49].
Discussion
Previous studies have established the dog as a strong model for clinical, pharmacologic and genetic studies of human iUC. To improve the clinical utility of the model we sought to develop a better understanding of the genomic profile associated with canine iUC by identifying differentially expressed genes among canine tumors versus normal tissue. We, and others, had previously defined a mutation in the BRAF gene and encoded protein in over 80% of canine iUC (BRAFV595E), that corresponds to the common BRAFV600E variant observed in humans [29, 30]. This is in contrast to human iUC for which the same mutation is rare. It is, however, common in human metastatic melanoma, colon and thyroid cancer, and rare leukemias [50,51,52,53].
A total of 3588 differentially expressed genes were identified by RNASeq in 15 iUC canine tumors compared to five normal tissue samples. We analyzed the top 100 up- and down-regulated genes from two previous studies of seven and eleven canine iUC tumors, and identified 65 and 81% of the same genes, respectively [27, 28]. Dividing our data by tumor cluster, the top 100 genes from past studies are most similar to the results we observe in the BRAFV595E tumors; 67 and 83% identical dysregulated genes, respectively. In contrast, the BRAFwt cluster shares only 49 and 62% of dysregulated genes. This is likely due to the overabundance (up to 87%) of BRAFV595E mutations in canine iUC [29, 30], thus skewing all lumped results toward the pattern of BRAFV595E tumors.
A total of 178 DE genes appear in all three studies, ignoring BRAFV595E mutation status. Of these (Additional File 6), 29 are associated with activation of EGFR and/or FGFR1 gene products in varying types of cancer (FDR qvalue = 4.68e− 15 and 3.74e− 15, MutsigDB). EGFR and FGFR1 appear to work together to increase tumorgenicity in lung cancer, and active FGFR1 can increase resistance to EGFR targeted therapies [54]. In this study we observe that ERBB2 is overexpressed in all canine iUC tumors. Five of those had significant z-scores (z-score = 2.54–4.65, equivalent to p < 0.01), and the remainder had z-scores surpassing standard measures of significance (z-score- = 1.70–2.49, equivalent to p < 0.05). HER2, the gene product of ERBB2 is upregulated in 37% of human iUCs and, combined with EGFR, is associated with advanced disease stage at diagnosis and increased risk of recurrence [55]. HER2 overexpression has been identified by immunohistochemistry in 57% of canine iUCs [56] and is predicted to be an upstream regulator of gene expression in canine iUCs [28]. Some growth factors, including EGFR and ERBB2, are the target of directed therapies for the treatment of iUCs in humans [57]. These findings suggest that canine iUC is an excellent platform for expanding our foundational understanding of growth factor receptor-targeted therapies. Clinical EGFR inhibitor trials are currently underway in dogs [3, 58].
We did not observe clustering based on luminal or basal signatures. Rather, all tumors in this dataset appear to have a luminal-like expression signature based on gene sets developed to distinguish these two molecular subtypes (Additional File 3; Supplementary Figs. 4A and B) [15, 26]. Our gene enrichment analysis demonstrates that all tumors in this study have an excess of dysregulated genes associated with human luminal-type tumors (pvalues = 7e− 83 and 3e− 125)(Additional File 7). Although not evident from the enrichment analysis, six tumors from the BRAFV595E cluster show upregulation of genes that are commonly used to identify the basal expression signature in human tumors in addition to luminal-type tumor marker genes (Additional File 3; Supplementary Fig. 4C). This expression pattern might indicate a small mixed tumor cluster, similar to the one termed UroB by Sjodahl et al. [17] UroB tumors can show signatures of both luminal and basal subtypes and have been assigned to both tumor types [17].
In the dataset presented here the tumor clusters correlate perfectly with the presence or absence of the BRAFV595E mutation. It is not uncommon for a somatic mutation to be overrepresented in a single expression cluster. For instance, FGFR3 mutations are found primarily in the luminal-papillary cluster of human iUC tumors [20]. However, perfect concordance is unusual given tumor heterogeneity. The exact concordance observed in this study is likely due to small numbers of tumors, and future analyses may reveal an overrepresentation of the BRAFV595E mutation in a single canine tumor cluster, much as is observed human studies. It is possible that non-conforming tumors may represent early stage tumor development in which the BRAFV595E mutation is not yet prevalent, or they may harbor unidentified somatic changes that activate the same pathways. Further investigation of BRAFwt tumors may reveal early mutations that initiate tumorigenesis, making them particularly good targets for therapy.
In this study, we included tumors from several dog breeds with an established increased risk of iUC. Nine out of the eleven BRAFV595E tumors were from four (Scottish Terrier, West Highland White Terrier, Shetland Sheepdog and Beagle) of the top seven high-risk breeds [4]. Only one of the samples in the BRAFwt cluster was from a high-risk breed. While the increased risk suggests a genetic predisposition to iUC, this is the first data to suggest that there may be common inherited mutations which may contribute to somatic mutation and expression patterns.
Analysis of the BRAFwt tumors shows an increase in immune activity, with 24 immune system-associated genes significantly upregulated compared to observations in both normal tissues as well as BRAFV595E tumors. This includes statistically significant enrichment of genes associated with interferon response and cytokine signaling pathways, and multiple upstream cytokine regulators (Fig. 2, Additional Files 5 and 8). Immune system gene signatures in human bladder tumors are considered to be associated with the infiltration of immune cells, i.e., not tumor cells [14, 17, 20]. Human tumors with strong evidence of immune infiltration have responded better to immune checkpoint therapies in some clinical trials [59]. Additionally, luminal infiltrated tumors respond well to radiation therapy, possibly because radiation triggers an immune response [60]. CCR5, one of the immune related genes upregulated in BRAFwt tumors is also upregulated in some human mammary tumors and appears to promote metastasis [61]. Antagonists of CCR5 are currently being assessed for their anti-tumor activity in aggressive tumors that express the gene [61,62,63].
Positional analysis of expression data highlights syntenic human genome regions that harbor common somatic copy number variants important in tumor development. Ten chromosomal regions in our study correspond to sites of recurring copy number amplification in a study of 11 different human tumor types, including bladder cancer [44]. Four of these occur on canine chromosome 13. Fluorescence in situ hybridization analysis reveals amplification of the entirety of chromosome 13 in canine iUCs [1], as does the expression level of individual genes in the region [27]. Our results highlight five regions on chromosome 13. The largest is 2.5 Mbs in the BRAFwt tumors and 1.5 Mbs in BRAFV595E (pvalue = 1− 21 and 3− 10 respectively). This region corresponds to human chromosome 8q24.3. This locus is amplified frequently in multiple human tumor types including those of the bladder and prostate [46, 64] Lymphocyte antigen 6 family member K (LY6K) is a gene associated with cell growth and is upregulated in human cell lines displaying 8q24.3 amplification [46]. In canine tumors, we observe that LY6E is amplified in BRAFwt tumors but in only half of BRAFV595E tumors. Genes at 8q24 have also been linked to MYC activity, which is located 10Mbs upstream of the amplified region. There is no significant change in expression of MYC between canine tumors and normal tissues in either cluster. However, pathway analysis predicts that MYC is activated as an upstream regulator of transcription in the BRAFV595E tumors (activation z-score = 4.4, p = 8.3e-6) but not in the BRAFwt tumors. These findings suggest there are different functional outcomes related to amplification at the 8q24.3 locus within canine iUCs. Thus, a comparison of canine and human syntenic regions could help define the functional and disease-relevant variants in loci involving large structural variants.
Of the four additional regions that display significantly upregulated groups of genes on canine chromosome 13 three are frequently amplified in human tumors. The first is located at 1.5 to 2.5 Mb and corresponds to human 8q22.2, which is duplicated in human bladder cancers [65, 66], while the second and third regions, from 43 to 44 Mb and 45.2 to 46.2 Mb, correspond to human 4q12, which is amplified in numerous human tumor types. The last locus harbors a cluster of over-expressed genes located between 57.7 and 58.7 Mb. Genes showing increased expression in this region include three different transmembrane protease serine 11 genes (TMPRSS11d, e and g). TMPRSS11e is associated with decreased survival in iUC patients [67, 68] and has been identified as a primary hub gene in co-expression networks marking iUC tumor progression [69]. This family of transmembrane proteins has not been extensively studied in iUC and, based on these results, is worth further investigation.
Our data demonstrates that the nectin4 gene is highly upregulated in BRAFwt tumors. Nectin4 lies in a region syntenic to human chromosome 1q23.3, and is overexpressed in human bladder and breast tumors, highlighting it’s potential as a drug target for epithelial cancers [49, 70]. A phase 1 clinical trial in humans has accomplished a 40% response rate with an antibody-drug conjugate targeting nectin4, and phase two and three trials are underway [71]. Although we predict that the entire locus is amplified in BRAFwt tumors, nectin4 is expressed above expected levels in all of the canine iUC tumors analyzed here, highlighting yet another clinical pathway in which studies of canine iUC could play a role in human treatment development.
Conclusions
In this study we examined expression patterns in BRAFV595E and BRAFwt tumors compared to normal tissue samples. We find distinct patters of dysregulated genes, with clusters of over and under-expressed genes limited to a small number of the dog’s 38 chromosomes. Notably, many of these regions highlight syntenic regions in the human genome that are associated with initiation or progression of human tumors, as well as long term and often deleterious outcomes. Future studies that include whole genome sequencing of large numbers of tumor/normal pairs will permit analysis of genotype/phenotype correlations.
The domestic dog is increasingly under consideration as a genetic system for the study of human disorders with underlying genetic components. In this study we further enhance that claim, demonstrating many molecular similarities in expression between human and dog iUCs. Our identification of these two tumor types in canine iUC has allowed us to identify new similarities between human and canine tumors and validate previously established observations. The fact that > 85% of canine tumors harbor BRAFV595E mutations which are correlated with expression patterns provides us with a mechanism to parse canine tumors in a way that is absent in humans, permitting early sub set analysis and improved matching for clinical trials of tumors with likely differing clinical outcomes. Further, our demonstration of expression profiles highly associated with somatic genotypes outlines distinct avenues to be explored for furthering our understanding of the underlying tumor biology of iUCs.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the NCBI short-read archive (SRA) under BioProject ID PRJNA559406 and PRJNA308949. https://www.ncbi.nlm.nih.gov/sra
Abbreviations
- iUC:
-
invasive urothelial carcinoma
- RFLP:
-
Restriction fragment length polymorphism
- TPM:
-
Transcripts per million
- SRA:
-
Short read archive
- MDS:
-
Multidimensional scaling
- PCA:
-
Principal component analysis
- FDR:
-
False discovery rate
- DE:
-
Differentially expressed
- Mbs:
-
Megabases
References
Shapiro SG, Raghunath S, Williams C, Motsinger-Reif AA, Cullen JM, Liu T, et al. Canine urothelial carcinoma: genomically aberrant and comparatively relevant. Chromosom Res. 2015;23(2):311–31.
American Society of Clinical Oncology. 2019. https://www.cancer.net/cancer-types/bladder-cancer/statistics. .
Sommer BC, Dhawan D, Ratliff TL, Knapp DW. Naturally-occurring canine invasive urothelial carcinoma: a model for emerging therapies. Bladder Cancer. 2018;4(2):149–59.
Knapp DW, Ramos-Vara JA, Moore GE, Dhawan D, Bonney PL, Young KE. Urinary bladder cancer in dogs, a naturally occurring model for cancer biology and drug development. ILAR J. 2014;55(1):100–18.
Charney VA, Miller MA, Heng HG, Weng HY, Knapp DW. Skeletal metastasis of canine urothelial carcinoma: pathologic and computed tomographic features. Vet Pathol. 2017;54(3):380–6.
Meuten DJ, editor. Tumors in domestic animals. 4th ed. Ames, Iowa: Blackwell Publishing; 2002.
Fulkerson CM, Knapp DW. Management of transitional cell carcinoma of the urinary bladder in dogs: a review. Vet J. 2015;205(2):217–25.
Vilar FO, de Araujo LA, Lima SV. Total bladder replacement with de-epithelialized ileum. Experimental study in dogs. Int Braz J Urol. 2004;30(3):237–44.
Knapp D, Glickman N, DeNicola D, Bonney P, Lin T, Glickman L. Naturally-occurring canine transitional cell carcinoma of the urinary bladder: a relevant model of human invasive bladder cancer. Urol Oncol. 2000;5:47–59.
Fulkerson CM, Dhawan D, Ratliff TL, Hahn NM, Knapp DW. Naturally occurring canine invasive urinary bladder cancer: a complementary animal model to improve the success rate in human clinical trials of new cancer drugs. Int J Genomics. 2017;2017:6589529.
Dhawan D, Ramos-Vara JA, Naughton JF, Cheng L, Low PS, Rothenbuhler R, et al. Targeting folate receptors to treat invasive urinary bladder cancer. Cancer Res. 2013;73(2):875–84.
Szigetvari NM, Dhawan D, Ramos-Vara JA, Leamon CP, Klein PJ, Ruple AA, et al. Phase I/II clinical trial of the targeted chemotherapeutic drug, folate-tubulysin, in dogs with naturally-occurring invasive urothelial carcinoma. Oncotarget. 2018;9(97):37042–53.
Hahn NM, Bonney PL, Dhawan D, Jones DR, Balch C, Guo Z, et al. Subcutaneous 5-azacitidine treatment of naturally occurring canine urothelial carcinoma: a novel epigenetic approach to human urothelial carcinoma drug development. J Urol. 2012;187(1):302–9.
Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25(2):152–65.
Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. 2014;111(8):3110–5.
Sjodahl G, Lauss M, Lovgren K, Chebil G, Gudjonsson S, Veerla S, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. 2012;18(12):3377–86.
Sjodahl G, Eriksson P, Liedberg F, Hoglund M. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J Pathol. 2017;242(1):113–25.
Aine M, Eriksson P, Liedberg F, Sjodahl G, Hoglund M. Biological determinants of bladder cancer gene expression subtypes. Sci Rep. 2015;5:10957.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22.
Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171(3):540–56 e25.
Eriksson P, Aine M, Veerla S, Liedberg F, Sjodahl G, Hoglund M. Molecular subtypes of urothelial carcinoma are defined by specific gene regulatory systems. BMC Med Genet. 2015;8:25.
Bertz S, Eckstein M, Stoehr R, Weyerer V, Hartmann A. Urothelial bladder cancer: An update on molecular pathology with clinical implications. Eur Urol Suppl. 2017;16(12):272–94.
Hussain SA, Palmer DH, Syn WK, Sacco JJ, Greensmith RM, Elmetwali T, et al. Gene expression profiling in bladder cancer identifies potential therapeutic targets. Int J Oncol. 2017;50(4):1147–59.
Seiler R, Ashab HAD, Erho N, van Rhijn BWG, Winters B, Douglas J, et al. Impact of molecular subtypes in muscle-invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur Urol. 2017;72(4):544–54.
Dhawan D, Paoloni M, Shukradas S, Choudhury DR, Craig BA, Ramos-Vara JA, et al. Comparative gene expression analyses identify luminal and basal subtypes of canine invasive urothelial carcinoma that mimic patterns in human invasive bladder cancer. PLoS One. 2015;10(9):e0136688.
Dhawan D, Hahn NM, Ramos-Vara JA, Knapp DW. Naturally-occurring canine invasive urothelial carcinoma harbors luminal and basal transcriptional subtypes found in human muscle invasive bladder cancer. PLoS Genet. 2018;14(8):e1007571.
Ramsey SA, Xu T, Goodall C, Rhodes AC, Kashyap A, He J, et al. Cross-species analysis of the canine and human bladder cancer transcriptome and exome. Genes Chromosomes Cancer. 2017;56(4):328–43.
Maeda S, Tomiyasu H, Tsuboi M, Inoue A, Ishihara G, Uchikai T, et al. Comprehensive gene expression analysis of canine invasive urothelial bladder carcinoma by RNA-Seq. BMC Cancer. 2018;18(1):472.
Decker B, Parker HG, Dhawan D, Kwon EM, Karlins E, Davis BW, et al. Homologous mutation to human BRAF V600E is common in naturally occurring canine bladder cancer-evidence for a relevant model system and urine-based diagnostic test. Mol Cancer Res. 2015;13(6):993–1002.
Mochizuki H, Kennedy K, Shapiro SG, Breen M. BRAF mutations in canine cancers. PLoS One. 2015;10(6):e0129534.
Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401–9.
Bell GI, Karam JH, Rutter WJ. Polymorphic DNA region adjacent to the 5′ end of the human insulin gene. Proc Natl Acad Sci U S A. 1981;78:5759–63.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006.
Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9.
Wucher V, Legeai F, Hedan B, Rizk G, Lagoutte L, Leeb T, et al. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017;45(8):e57.
Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–3.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Menche J, Guney E, Sharma A, Branigan PJ, Loza MJ, Baribaud F, et al. Integrating personalized gene expression profiles into predictive disease-associated gene pools. NPJ Syst Biol Appl. 2017;3:10.
Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131(4):281–5.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40.
Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30(4):523–30.
Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10):1134–40.
de Matos SR, Dalleau S, Williamson KE, Emmert-Streib F. Urothelial cancer gene regulatory networks inferred from large-scale RNAseq, Bead and Oligo gene expression data. BMC Syst Biol. 2015;9:21.
Matsuda R, Enokida H, Chiyomaru T, Kikkawa N, Sugimoto T, Kawakami K, et al. LY6K is a novel molecular target in bladder cancer on basis of integrate genome-wide profiling. Br J Cancer. 2011;104(2):376–86.
Lopez V, Gonzalez-Peramato P, Suela J, Serrano A, Algaba F, Cigudosa JC, et al. Identification of prefoldin amplification (1q23.3-q24.1) in bladder cancer using comparative genomic hybridization (CGH) arrays of urinary DNA. J Transl Med. 2013;11:182.
Riester M, Werner L, Bellmunt J, Selvarajah S, Guancial EA, Weir BA, et al. Integrative analysis of 1q23.3 copy-number gain in metastatic urothelial carcinoma. Clin Cancer Res. 2014;20(7):1873–83.
Challita-Eid PM, Satpayev D, Yang P, An Z, Morrison K, Shostak Y, et al. Enfortumab vedotin antibody-drug conjugate targeting Nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016;76(10):3003–13.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54.
Boulalas I, Zaravinos A, Delakas D, Spandidos DA. Mutational analysis of the BRAF gene in transitional cell carcinoma of the bladder. Int J Biol Markers. 2009;24(1):17–21.
Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6(4):313–9.
Jung CK, Little MP, Lubin JH, Brenner AV, Wells SA Jr, Sigurdson AJ, et al. The increase in thyroid cancer incidence during the last four decades is accompanied by a high frequency of BRAF mutations and a sharp increase in RAS mutations. J Clin Endocrinol Metab. 2014;99(2):E276–85.
Quintanal-Villalonga A, Molina-Pinelo S, Cirauqui C, Ojeda-Marquez L, Marrugal A, Suarez R, et al. FGFR1 cooperates with EGFR in lung cancer oncogenesis, and their combined inhibition shows improved efficacy. J Thorac Oncol. 2019;14(4):641–55.
Li W, Wang Y, Tan S, Rao Q, Zhu T, Huang G, et al. Overexpression of epidermal growth factor receptor (EGFR) and HER-2 in bladder carcinoma and its association with patients' clinical features. Med Sci Monit. 2018;24:7178–85.
Millanta F, Impellizeri J, McSherry L, Rocchigiani G, Aurisicchio L, Lubas G. Overexpression of HER-2 via immunohistochemistry in canine urinary bladder transitional cell carcinoma - a marker of malignancy and possible therapeutic target. Vet Comp Oncol. 2018;16(2):297–300.
Sjodahl G, Jackson CL, Bartlett JM, Siemens DR, Berman DM. Molecular profiling in muscle-invasive bladder cancer: more than the sum of its parts. J Pathol. 2019;247(5):563–73.
Nagaya T, Okuyama S, Ogata F, Maruoka Y, Knapp DW, Karagiannis SN, et al. Near infrared photoimmunotherapy targeting bladder cancer with a canine anti-epidermal growth factor receptor (EGFR) antibody. Oncotarget. 2018;9(27):19026–38.
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–20.
Efstathiou JA, Mouw KW, Gibb EA, Liu Y, Wu CL, Drumm MR, et al. Impact of immune and stromal infiltration on outcomes following bladder-sparing trimodality therapy for muscle-invasive bladder cancer. Eur Urol. 2019;76(1):59–68.
Velasco-Velazquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, et al. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012;72(15):3839–50.
Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell. 2016;29(4):587–601.
Pervaiz A, Zepp M, Mahmood S, Ali DM, Berger MR, Adwan H. CCR5 blockage by maraviroc: a potential therapeutic option for metastatic breast cancer. Cell Oncol (Dordr). 2019;42(1):93–106.
Saramaki OR, Porkka KP, Vessella RL, Visakorpi T. Genetic aberrations in prostate cancer by microarray analysis. Int J Cancer. 2006;119(6):1322–9.
Hu Y, Xing J, Wang L, Huang M, Guo X, Chen L, et al. RGS22, a novel cancer/testis antigen, inhibits epithelial cell invasion and metastasis. Clin Exp Metastasis. 2011;28(6):541–9.
Veltman JA, Fridlyand J, Pejavar S, Olshen AB, Korkola JE, DeVries S, et al. Array-based comparative genomic hybridization for genome-wide screening of DNA copy number in bladder tumors. Cancer Res. 2003;63(11):2872–80.
Human Protein Atlas. 2019. v18.1. www.proteinatlas.org/ENSG00000087128-TMPRSS11E/pathology/tissue/urothelial+cancer. Accessed 21 Sept 2019.
Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352). https://doi.org/10.1126/science.aan2507.
Zhang DQ, Zhou CK, Chen SZ, Yang Y, Shi BK. Identification of hub genes and pathways associated with bladder cancer based on co-expression network analysis. Oncol Lett. 2017;14(1):1115–22.
Targeting Nectin-4 in bladder cancer [press release]. Aug 2017.
Alhalabi O, Rafei H, Shah A, Siefker-Radtke A, Campbell M, Gao J. Targeting advanced urothelial carcinoma-developing strategies. Curr Opin Oncol. 2019;31(3):207–15.
Acknowledgements
We thank the many dog owners who allowed their pets to participate in this study. We acknowledge the work of the NIH Intramural Sequencing Center for producing the next-generation sequencing data.
Funding
This work was funded by the Intramural Program of the National Human Genome Research Institute at NIH, AKC-Canine Health Foundation Grants 754, 1336, and 1577 (to E.A.O., H.G.P. and D.W.K.), and private donations made for bladder cancer research at Purdue University (D.W. K.).
Author information
Authors and Affiliations
Contributions
HGP performed laboratory experiments, analyzed and interpreted genomic data, and wrote the manuscript. DD provided biological samples and data for replication. ACH preformed laboratory experiments. JAR-V performed pathology on specimens. BWD contributed to bioinformatic analysis. DWK provided samples, analyzed patient data, provided suggestions on analysis and contributed to manuscript writing. EAO oversaw all laboratory and analytical experiments and wrote the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Sample collection was performed following approval of the Animal Care and Use Committees of the collecting institutions, and owners of all participating dogs signed an informed consent document.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:.
Figures S1–S4. Supporting figures for results.
Additional file 2:.
Table S1. Expression values of Ensembl genes.
Additional file 3:.
Table S2. Expression values of predicted non-coding genes.
Additional file 4:.
Table S3. Genes that are differentially expressed between BRAFV595E and BRAFwt tumors.
Additional file 5:.
Table S4. Gene groups overrepresented in the genes that distinguish BRAFV595E tumors from BRAFwt tumors.
Additional file 6:.
Table S5. Comparison of gene expression values from previously published datasets.
Additional file 7:.
Table S6. Curated gene sets over-represented in canine iUC.
Additional file 8:.
Table S7. Hallmark gene sets over-represented in canine iUC.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Parker, H.G., Dhawan, D., Harris, A.C. et al. RNAseq expression patterns of canine invasive urothelial carcinoma reveal two distinct tumor clusters and shared regions of dysregulation with human bladder tumors. BMC Cancer 20, 251 (2020). https://doi.org/10.1186/s12885-020-06737-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-06737-0