
Abstract
Background
Non-small cell lung cancer (NSCLC) as the most frequent type of lung cancer is associated with extensive mortality. Researchers have studied the suitability of several molecules as biomarkers for early detection of this cancer. Long non-coding RNAs (lncRNAs) as the main regulators of gene expression have also been assessed in this regard.
Methods
In the present study, we compared expression level of Fas-antisense 1 (FAS-AS1), Growth Arrest Specific 5 (GAS5), PVT1, Nuclear Paraspeckle Assembly Transcript 1 (NEAT1), HOXA transcript antisense RNA myeloid-specific 1 (HOTAIRM1), taurine upregulated gene 1 (TUG1) and TNFα and hnRNPL related immunoregulatory LincRNA (THRIL) in 32 NSCLC samples and their corresponding adjacent non-cancerous tissues (ANCTs).
Results
NEAT1 has been significantly over-expressed in NSCLC tissues obtained from male subjects compared with the corresponding ANCTs (Relative expression (REx) = 3.022, P = 0.019) but not in female subjects (P = 0.975). FAS-AS1 was significantly down-regulated in NSCLC tissues obtained from both males and females subjects compared with the corresponding ANCTs (REx = − 4.12 and − 3.14, P = 0.015 and 0.033 respectively). TUG1, GAS5, THRIL and HOTAIRM1 were significantly down-regulated in tumoral tissues obtained from male subjects compared with the corresponding ANCTs.
Conclusions
The observed dysregulation of these lncRNAs in NSCLC tissues compared with the corresponding ANCTs warrants future studies to confirm the results of the current study in larger sample sizes to elaborate their role as cancer biomarkers.
Similar content being viewed by others

Background
Lung cancer as the most frequent malignancy and the foremost source of cancer mortality is a heterogeneous disorder. The most common type of lung cancer is non-small-cell lung cancer (NSCLC) which accounts for 85% of the total cases and is further classified into adenocarcinoma, large cell carcinoma and squamous cell carcinoma subtypes [1]. Collectively two thirds of patients with NSCLC are being diagnosed when the tumor is locally advanced or has metastasized [2]. Such delay in the diagnosis of lung cancer in addition to the absence of appropriate therapeutic targets lead to poor patients’ outcome [3]. Consequently, researchers invested substantial efforts in the identification of diagnostic biomarkers and therapeutic targets for this type of human malignancy. Among these putative biomarkers are long non-coding RNAs (lncRNAs) [3]. This proportion of human genome plays fundamental roles in the regulation of tumor suppressor genes and oncogenes expression via epigenetic, transcriptional, and post-transcriptional mechanism [4] and is dysregulated in several human malignancies including NSCLC [5]. A comprehensive study in lung adenocarcinoma has led to identification of 2420 lncRNAs with significant differential expression between tumor and normal tissue samples [6]. Moreover, in silico analysis of NSCLC expression profiles in the Gene Expression Omnibus (GEO) has resulted in recognition of 47 dysregulated lncRNAs in these patients [7]. In addition, dysregulation of lncRNAs in lung cancer tissues has been associated with air pollution [8]. Some well-known risk factors for NSCLC also trigger expression of lncRNAs such as the smoke and cancer–associated lncRNA–1 (SCAL1), DQ786227, and LOC728228 in these tissues [3]. Notably, Wu et al. have detected subtype-dependent lncRNA-associated protein-protein interaction (PPI) modules in human lung cancer and proposed distinct molecular mechanisms for every single subtype. They also demonstrated functional link between antisense lncRNAs and sense genes [9]. Even low ample lncRNAs such as the so-called Viability Enhancing LUng Cancer Transcript (VELUCT) exert functional roles in the pathogenesis of lung cancer [10]. Other studies have demonstrated aberrant expression of a number lncRNAs including the Prostate cancer-associated transcript1 (PCAT1) [11], Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1) [12] and Cancer-Associated Region Long non-coding RNA (CARLo-5) [13] in NSCLC tissues and showed possible links between their expression and malignant features of these cells or patients’ outcomes.
In the present study, in an effort to evaluate the suitability of lncRNAs as biomarkers for NSCLC we compared expression level of seven apoptosis related lncRNAs namely Fas-antisense 1 (FAS-AS1), Growth Arrest Specific 5 (GAS5), PVT1, Nuclear Paraspeckle Assembly Transcript 1 (NEAT1), HOXA transcript antisense RNA myeloid-specific 1 (HOTAIRM1), taurine upregulated gene 1 (TUG1) and TNFα and hnRNPL related immunoregulatory LincRNA (THRIL) in 32 NSCLC samples and their corresponding adjacent non-cancerous tissues (ANCTs) and plotted the receiver operating characteristic (ROC) curve to estimate their appropriateness for classifying disease status. To the best of our knowledge, the current study is the first study to assess relative expression of HOTAIRM1, THRIL and FAS-AS1 in lung cancer tissues compared with ANCTs using the quantitative real-time PCR. NEAT1 is an apoptosis-related lncRNA with remarkable over-expression in plasma samples of NSCLC patients [14]. Contribution of GAS5 in the pathogenesis of lung cancer has been highlighted through the observed associations between genomic variants within this gene and risk of this malignancy [15]. TUG1 has been previously shown to exert a tumor suppressor role in NSCLC [16]. Finally, a previous study has suggested a role for PVT1 in the pathogenesis of NSCLC through inhibition of p15 and p21 expression [17].
In the current investigation, we also assessed the correlation between expression levels of these lncRNAs to find any possible similar regulatory mechanism for these lncRNAs in the context of lung cancer.
Methods
Patients’ samples
Cancer samples and the corresponding ANCTs were excised during surgery from 32 patients being admitted at Labbafinejad Hospital with definite diagnosis of NSCLC. None of patients received radiotherapy or chemotherapy before surgery. Tissue samples were transferred to laboratory of Medical Genetics Department in liquid nitrogen. Informed consent forms were obtained from all study participants. The study protocol was approved by the ethical committee of Shahid Beheshti University of Medical Sciences (IR.SBMU.MSP.REC.1395.525). In this study, all methods were performed in accordance with the relevant guidelines and regulations.
Sampling and RNA extraction
Total RNA was isolated from cancerous tissues and ANCTs using the TRIzol™ Reagent (Invitrogen, Carlsbad, CA, USA) according to the guidelines. The extracted RNA was supposed to DNase I treatment to get rid of DNA contamination. The quantity and quality of the extracted RNA was assessed by Nanodrop equipment (Thermo Scientific) and gel electrophoresis.
cDNA synthesis and quantitative RT-PCR
cDNA was synthetized from RNA samples using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit. The relative expression level of each lncRNA was compared between tumoral and non-tumoral tissues using the rotor gene 6000 Corbett Real-Time PCR System. HPRT1 was used as the reference gene. Primers and probes used for PCR were designed using the Allele ID 7 for × 64 windows software (Premier Biosoft, Palo Alto, USA). The primers and probes sequences and PCR product length are demonstrated in Table 1. Applied Biosystems TaqMan® Universal PCR Master Mix was used for quantification of lncRNAs expression. PCR program included a denaturation step at 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 60 s and a final extension step in 72 °C for 5 min.
Statistical analysis
Relative expression of lncRNAs in tumoral tissues compared with ANCTs was estimated based on calculation of Ln [Efficiency^∆CT] values. The association between lncRNAs transcript levels and clinicipathologic data of patients was evaluated using Chi-square test. Spearman rank order correlation test was used to estimate the correlation between relative expression levels of lncRNAs and patients’ age. Statistical analyses were performed in R 3.5.1. The effects of possible confounding variables such as age and sex with were assessed using the Quantile regression model. Differences between tumoral and ANCTs were analyzed using Bayesian modeling in RStan using brms and BEST package with Iteration = 5000 and Warmup = 2000. Convergence was assessed using Rhat parameter. P values less than 0.05 were considered significant.
The receiver operating characteristic (ROC) curve was plotted to evaluate the suitability of gene expression levels for classifying disease status. In order to estimate gene expression probability cut-off the Youden index (j) was used to maximize the difference between sensitivity (true-positive rate) and 1 – specificity (false-positive rate). The accuracy of each marker for diagnosis of lung cancer was scored based on the area under curve (AUC) values using the following system: 0.90–1 = excellent (A), 0.80–0.90 = good (B), 0.70–0.80 = fair (C), 0.60–0.70 = poor (D) and 0.50–0.60 = fail (F).
In silico analyses
We used LncRNAtor online tool [18] to assess target genes of lncRNAs in lung cancer tissues. The retrieved target genes were scored based on r and P values and those with r > 0.2 and P < 0.05 were subjected to further Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis by DAVID 6.8 tool (https://david.ncifcrf.gov/summary.jsp). Finally, we assessed lncRNAs targets at protein level by using starBase v2.0 [19]. The interaction network between theses lncRNAs and their targets was depicted using Gene MANIA tool [20].
Results
General clinical and demographic data of patients
The mean age of study participants was 57.96 ± 7.73 years, ranging from 37 to 80 years. Other features are shown in Table 2.
Relative expression of lncRNAs in tumoral tissues vs. ANCTs
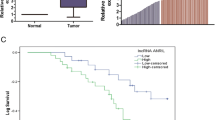
Among the lncRNAs, NEAT1 was the only up-regulated lncRNA in tumoral tissues while GAS5 had the highest down-regulation in tumoral tissues compared with ANCTs. NEAT1 has been significantly over-expressed in NSCLC tissues obtained from male subjects compared with the corresponding ANCTs (Relative expression (REx) = 3.022, P = 0.019) but not in female subjects (P = 0.975). FAS-AS1 was significantly down-regulated in NSCLC tissues obtained from both males and females subjects compared with the corresponding ANCTs (REx = − 4.12 and − 3.14, P = 0.015 and 0.033 respectively). TUG1, GAS5, THRIL and HOTAIRM1 were significantly down-regulated in tumoral tissues obtained from male subjects compared with the corresponding ANCTs (Table 3). Figure 1 shows relative expression of lncRNAs in tumor tissues and ANCTs.

Relative expression of lncRNAs in NSCLC samples and ANCTs
Association study of lncRNAs expression levels and clinicopathological data of patients
No significant association was found between expression levels of mentioned lncRNAs and patients’ clinicopathologic data when dividing patients into down−/up-regulation categories based on relative expression of each lncRNA in tumoral tissue compared with the paired ANCT (Table 4). However, a significant association was found between relative expression of TUG1 and cancer subtype (Table 5).
Correlation analysis between expression levels of lncRNAs in tumoral tissues and ANCTs
Spearman Correlation analysis revealed significant correlations between relative expression levels of lncRNAs especially within tumor tissues and in male subgroup (Table 6).
ROC curve analysis
Based on ROC curve analysis results, the accuracy of GAS5 expression levels for lung cancer diagnosis is good (Fig. 2). Besides, TUG1, FAS-AS1 and THRIL expression levels were fair diagnostic markers for lung cancer. Table 7 shows the details of ROC curve analysis.

ROC curve analysis for GAS5
We also combined all differentially expressed lncRNAs in ROC curve analysis. This method raised the diagnostic power to 0.898 based on the obtained AUC value (Fig. 3).

ROC curve analysis for combination of differentially expressed lncRNAs
KEGG pathway enrichment analysis
KEGG pathway enrichment analysis showed the targeted genes participate in a number of cancer-related pathways such as chemokine signaling, HIF-1, JAK-STAT and NOTH and thyroid hormone signaling pathways as well as some virus-associated pathways. Table 8 shows the results of KEGG pathway enrichment analysis.
GO analysis of differentially expressed target genes of lncRNAs in lung cancer
The lncRNAs target genes are involved in cancer-related cellular processes such as cell cycle control, cell division, translation and signal transduction (Table 9).
Finally, we provided a list of differentially expressed target proteins of lncRNAs in lung cancer using starBase tool (Table 10) and depicted the network between these lncRNAs and their targets (Fig. 4). The enriched pathways were related to gene silencing by RNA, regulation of translation, mRNA processing, RNA splicing and posttranscriptional regulation of gene expression.

Analysis of interaction network between these lncRNAs and their targets showed that the enriched pathways were related to gene silencing by RNA, regulation of translation, mRNA processing, RNA splicing and posttranscriptional regulation of gene expression
Discussion
Identification and characterization of novel diagnostic and prognostic biomarkers is expected to improve NSCLC patients’ outcomes. The tissue- or cell-specific expression profile of lncRNAs potentiates them as appropriate biomarkers in this regard [3]. In the present study, we evaluated expression pattern of seven lncRNAs in NSCLC samples and their matched ANCTs and showed a gender specific pattern of lncRNA dysregulation in tumoral tissues. NEAT1 has been significantly over-expressed in NSCLC tissues obtained from male subjects compared with the corresponding ANCTs but not in female subjects. NEAT1 has been among three lncRNAs with significant over-expression in plasma samples of NSCLC patients [14]. Moreover, NETA1 over-expression in NSCLC tissues has been demonstrated in a cohort of 125 patients with significant correlation between its expression levels and patient, lymphatic metastasis, vascular invasion and clinical TNM stage [21]. Our data is in line with the results of these two studies in the terms of NEAT1 over-expression. However, lack of correlation between expression levels of this lncRNA and clinicopathologic data of patients can be at least partly explained by the relative small sample size of the current study.
We also detected significant down-regulation of FAS-AS1 in NSCLC tissues obtained from both males and females subjects compared with the corresponding ANCTs. This lncRNA has an inhibitory role in alternative splicing of Fas to produce soluble Fas receptor (sFas) in lymphomas. Ectopic expression of FAS-AS1 leading to down-regulation of sFas has been suggested as a treatment modality in lymphoma [22]. Although the function of this lncRNA has not been assessed in lung cancer cells yet, a previous study has shown the co-expression of Fas and Fas ligand (FasL) in lung cancer cell lines and the apoptotic effect of agonistic anti-Fas antibody in these cells [23]. Future studies are needed to explain the role and status of FAS-AS1 in regulation of Fas in lung cancer cells.
Moreover, we demonstrated significant down-regulation of TUG1, GAS5, THRIL and HOTAIRM1 in tumoral tissues obtained from male subjects compared with the corresponding ANCTs. TUG1 down-regulation has been recently demonstrated in NSCLC tissues obtained from Taiwanese patients [24]. More importantly, they observed a more significant down-regulation of this lncRNA in samples obtained from male patients [24] which is in accordance with our data. GAS5 has been regarded as a tumor suppressor in NSCLC whose expression was significantly lower in tumoral tissues compared with ANCTs. Such down-regulation has been correlated with TNM stage but not tumor size, lymph node metastasis, age, gender, differentiation and histology type in NSCLC [25]. Consequently, our data regarding gender-specific down-regulation of GAS5 is not supported by the result of this study. THRIL is an lncRNA with regulatory role on TNFα expression and the consequent innate immune response [26]. Although the role of this lncRNA in carcinogenesis has not elaborated yet, the observed down-regulation of it in NSCLC warrants future studies to explain its participation in this kind of human malignancy. Finally, HOTAIRM1 is a principal regulator of myeloid cell development by targeting HOXA1. HOTAIRM1 over-expression in myeloid-derived suppressor cells (MDSCs) results in down-regulation of the expression of suppressive molecules in these cells. On the other hand, HOTAIRM1 levels were shown to be down-regulated in the peripheral blood cells of lung cancer patients compared to those of healthy controls [27]. Consequently, the observed down-regulation of this lncRNA in tumoral tissues of male patients is in line with the previous studies regarding the role of this lncRNA in the pathogenesis of cancer.
Although we assessed expression profile of some lncRNAs in NSCLC using quantitative real time PCR, it is anticipated that computational modeling would be used in near future for the identification of potential NSCLC-related lncRNAs or microRNAs. Computational models would facilitate selection of the most promising candidates for further laboratory investigation so decreasing the labor of the biological researches [28]. The availability of lncRNA-related databases such as those demonstrating annotation of lncRNAs sequences or structures as well as the experimentally validated lncRNA–disease associations or interactions has facilitated this process [29]. Perhaps one of the most important features of these computational models for detection of possible disease-related lncRNAs is possibility of application of a certain model in similar disorders as similar diseases are expected to be linked with functionally comparable lncRNAs [30]. Two recently developed tools for prediction of novel miRNA-disease associations have been shown to be effective and powerful tools for such propose in a wide range of human malignancies [31, 32].
In addition, we demonstrated significant correlations between relative expression levels of lncRNAs especially within tumor tissues and in male subgroup. Such correlations might imply the presence of a single regulatory mechanism for expression of these lncRNAs. Future studies are needed to clarify such mechanism. We also assessed the accuracy of expression levels of these genes in lung cancer diagnosis and demonstrated the best values for GAS5. By plotting ROC curves to evaluate the ability of lncRNAs expression to improve the prediction of lung cancer, GAS5 transcript levels had more than 80% specificity and sensitivity in this regard. On the other hand, TUG1, FAS-AS1, HOTAIRM1 and THRIL have been demonstrated to be specific markers despite their low sensitivity. Based on these results we recommend future evaluation of this panel of markers in larger samples sizes of NSCLC patients.
Finally, we evaluated target genes of these lncRNAs at both mRNA and protein levels in lung cancer using online tools. We demonstrated involvement of these targets in a number of molecular/signaling networks most of them being recognized as cancer hallmarks. Most importantly, the interactive network between lncRNAs and their targets was shown to participate in different aspects of expression regulation including gene silencing by RNA, regulation of translation, mRNA processing, RNA splicing and posttranscriptional regulation of gene expression.
Conclusions
In brief, in the present study we demonstrated dysregulation of seven lncRNAs in NSCLC tissues compared with the corresponding ANCTs. Such observations underscore the role of these lncRNAs in the pathogenesis of lung cancer and suggest them as possible biomarkers for this malignancy. Future studies are needed to confirm the results of the current study in larger sample sizes to elaborate their role as cancer biomarkers.
Abbreviations
- ANCTs:
-
Adjacent non-cancerous tissues
- AUC:
-
Area under curve
- FAS-AS1:
-
Fas-antisense 1
- GAS5:
-
Growth Arrest Specific 5
- GO:
-
Gene Ontology
- HOTAIRM1:
-
HOXA transcript antisense RNA myeloid-specific 1
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- NEAT1:
-
PVT1, Nuclear Paraspeckle Assembly Transcript 1
- NSCLC:
-
Non-small cell lung cancer
- ROC:
-
Receiver operating characteristic
- THRIL:
-
TNFα and hnRNPL related immunoregulatory LincRNA
- TUG1:
-
taurine upregulated gene 1
References
Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male : female differences diminishing and adenocarcinoma rates rising. Int J Cancer. 2005;117(Nov 1):294.
Morgensztern D, Ng SH, Gao F, Govindan R. Trends in stage distribution for patients with non-small cell lung cancer: a National Cancer Database survey. J Thorac Oncol. 2010;5:29.
Wei MM, Zhou GB. Long non-coding RNAs and their roles in non-small-cell lung Cancer. Genom Proteom Bioinf. 2016;14:280.
Taheri M, Omrani MD, Ghafouri-Fard S. Long non-coding RNA expression in bladder cancer. Biophys Rev. 2018;10(4):1205–13.
E. Farbod et al., Expression analysis of OIP5-AS1 in non-small cell lung Cancer. Klin Onkol 31, 260 (Summer, 2018).
Xu G, et al. Long noncoding RNA expression profiles of lung adenocarcinoma ascertained by microarray analysis. PLoS One. 2014;9(8):e104044.
Yang JC, et al. Analysis of lncRNA expression profiles in non-small cell lung cancers (NSCLC) and their clinical subtypes. Lung Cancer. 2014;85:110.
Wei MM, et al. Long non-coding RNA stabilizes the Y-box-binding protein 1 and regulates the epidermal growth factor receptor to promote lung carcinogenesis. Oncotarget. 2016;7:59556.
Wu CH, Hsu CL, Lu PC, Lin WC, Juan HF, Huang HC. Identification of lncRNA functions in lung cancer based on associated protein-protein interaction modules. Sci Rep-Uk. 2016;6:35939.
Seiler J, et al. The lncRNA VELUCT strongly regulates viability of lung cancer cells despite its extremely low abundance. Nucleic Acids Res. 2017;45:5458.
Zhao B, Hou XB, Zhan H. Long non-coding RNA PCAT-1 over-expression promotes proliferation and metastasis in non-small cell lung cancer cells. Int J Clin Exp Med. 2015;8:18482.
Zhu LC, Liu JH, Ma SL, Zhang SR. Long noncoding RNA MALAT-1 can predict metastasis and a poor prognosis: a meta-analysis. Pathol Oncol Res. 2015;21:1259.
Luo J, et al. Long non-coding RNA CARLo-5 is a negative prognostic factor and exhibits tumor pro-oncogenic activity in non-small cell lung cancer. Tumor Biol. 2014;35:11541.
Hu XD, et al. The plasma lncRNA acting as fingerprint in non-small-cell lung cancer. Tumor Biol. 2016;37:3497.
Li WH, et al. Genetic variation of lncRNA GAS5 contributes to the development of lung cancer. Oncotarget. 2017;8:91025.
Zhang E, et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. 2014;5:e1243.
Cui D, et al. Long non-coding RNA PVT1 as a novel biomarker for diagnosis and prognosis of non-small cell lung cancer. Tumour Biol. 2016;37:4127.
Park C, Yu N, Choi I, Kim W, Lee S. lncRNAtor: a comprehensive resource for functional investigation of long non-coding RNAs. Bioinformatics. 2014;30:2480.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92.
Warde-Farley D, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214.
Pan LJ, et al. Upregulation and clinicopathological significance of long non-coding NEAT1 RNA in NSCLC tissues. Asian Pac J Cancer Prev. 2015;16:2851.
Sehgal L, et al. FAS-antisense 1 lncRNA and production of soluble versus membrane FAS in B-cell lymphoma. Leukemia. 2014;28:2376.
Kawasaki M, et al. Analysis of Fas and Fas ligand expression and function in lung cancer cell lines. Eur J Cancer (Oxford, England : 1990). 2000;36:656.
Lin PC, Huang HD, Chang CC, Chang YS, Yen JC, Lee CC, Chang WH, Liu TC, Chang JG. Long noncoding RNA TUG1 is downregulated in non-small cell lung cancer and can regulate CELF1 on binding to PRC2. BMC Cancer. 2016;16(1):583.
Shi XF, et al. A critical role for the long non-coding RNA GAS5 in proliferation and apoptosis in non-small-cell lung cancer. Mol Carcinogen. 2015;54:E1.
Li Z, et al. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci U S A. 2014;111:1002.
Tian X, et al. LncRNA HOTAIRM1-HOXA1 axis down-regulates the immunosuppressive activity of myeloid-derived suppressor cells in lung cancer. Front Immunol. 2018;9:473.
Chen X, Xie D, Zhao Q, You ZH. MicroRNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2017.
Chen X, Yan CC, Zhang X, You ZH. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2017;18:558.
Chen X, Yan GY. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics. 2013;29:2617.
Chen X, Huang L. LRSSLMDA: Laplacian regularized sparse subspace learning for MiRNA-disease association prediction. PLoS Comput Biol. 2017;13:e1005912.
You ZH, et al. PBMDA: a novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput Biol. 2017;13:e1005455.
Acknowledgements
The present study was supported by a grant from Shahid Beheshti University of Medical Sciences (grant number: 12810).
Ethics approval and consent to participant
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent forms were obtained from all study participants. The study protocol was approved by the ethical committee of Shahid Beheshti University of Medical Sciences (IR.SBMU.MSP.REC.1395.525). All methods were performed in accordance with the relevant guidelines and regulations.
Funding
Not applicable.
Availability of data and materials
The analysed data sets generated during the study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
SGF wrote the manuscript and contributed in study design. MT and FE supervised the study and performed the experiment. SAJ analyzed the data. MBS was the clinical consultant and assessed patients for inclusion in the study. RS and MDO conducted the bioinformatics analyses. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Esfandi, F., Taheri, M., Omrani, M.D. et al. Expression of long non-coding RNAs (lncRNAs) has been dysregulated in non-small cell lung cancer tissues. BMC Cancer 19, 222 (2019). https://doi.org/10.1186/s12885-019-5435-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-019-5435-5