
Abstract
Background
Chimeric antigen receptor T (CAR T) cells immunotherapy is rapidly developed in treating cancers, especially relapsed or refractory B-cell malignancies.
Methods
To assess the efficacy and safety of CAR T therapy, we analyzed clinical trials from PUBMED and EMBASE.
Results
Results showed that the pooled response rate, 6-months and 1-year progression-free survival (PFS) rate were 67%, 65.62% and 44.18%, respectively. We observed that received lymphodepletion (72% vs 44%, P = 0.0405) and high peak serum IL-2 level (85% vs 31%, P = 0.04) were positively associated with patients’ response to CAR T cells. Similarly, costimulatory domains (CD28 vs CD137) in second generation CAR T was positively associated with PFS (52.69% vs 33.39%, P = 0.0489). The pooled risks of all grade adverse effects (AEs) and grade ≥ 3 AEs were 71% and 43%. Most common grade ≥ 3 AEs were fatigue (18%), night sweats (14%), hypotension (12%), injection site reaction (12%), leukopenia (10%), anemia (9%).
Conclusions
In conclusion, CAR T therapy has promising outcomes with tolerable AEs in relapsed or refractory B-cell malignancies. Further modifications of CAR structure and optimal therapy strategy in continued clinical trials are needed to obtain significant improvements.
Similar content being viewed by others

Background
Recently, chimeric antigen receptor T (CAR T) cells immunotherapy is rapidly developed. Generally, CAR consists of tumor associated antigen (TAA) binding domain, hinge domain, transmembrane domain and signaling domain. TAA usually is a single-chain variable fragment (scFv). Unlike physiological T cell receptors (TCR), scFv can recognize antigen directly without major histocompatibility complex (MHC) restriction. Intracellular signaling domains generally contain immunoreceptor tyrosine-based activation motifs (ITAMs), which usually is CD3ζ and costimulatory molecule (CM), including CD28, CD134 (OX40) and CD137 (4-1BB) [1,2,3,4]. T cell activation is initiated through the ITAMs presented in the CD3 polypeptides [5]. The first generation of CAR contains a single signaling domain, usually are CD3ζ chain [6]. Second generation CAR have one signaling domain, and one costimulation domain, with which T cells can expand and functioning under the exist of antigen [1]. Three signaling domains with two costimulatory molecules were engineered to design the third generation CAR. CAR T therapy including the following procedures: first, collecting T cells from the patient or donor; second, isolating and activating T cells [7]; third, modifing T cells with CARs with viral vector transduction or electroporation of RNA or DNA; fourth, expanding the transduced cells; finally, patients receive lymphodepletion and the infusion (Fig. 1).

CAR T cell therapy. T lymphocytes from the patient or a suitable donor are isolated. Then T cells are activated with anti-human CD3/CD28 antibody-coated beads, anti-CD3 monoclonal antibodies, and/or artificial antigen-presenting cells(APCs). The first, second or third generation CARs are transducted to T cells via a viral or nonviral vector (i.e., eletroporation). Engineered CAR T cells are expanded and infused into the patient who received or not received lymphodepletion regimen
CD19 is expressed restrictively to B cells, so it is a potential target [8, 9]. CD20 exists in over 90% of B-cell lymphomas and is also used to treat non-Hodgkin’s lymphoma (NHL) [10, 11]. However, there were great difference of efficacy in different trials. Additionally, the efficacy of CAR T cells might be affected by the different execute procedures. However, the critical factors for better efficacy are still unclear.
The adverse effects of CAR T therapy were big challenges, including the cytokine release syndrome(CRS), on-target off-tumor toxicities and toxicities caused by the lymphodepletion chemotherapy [4, 12,13,14]. Fevers, fatigue and hypotension were often reported [4, 12,13,14]. However, the most frequently occurred events and the incidence of any treatment adverse events are unknown.
Previous study evaluated the efficacy of anti-CD19 CAR T cells therapy, but it didn’t assess the factors related to progression free survival and the safety of this therapy [15]. The two systematic reviews which estimate efficacy and safety of anti-CD19 CAR T cells therapy were limited because that only 5 and 6 trials were included, respectively [16, 17]. In this study, we aimed to assess the efficacy and safety of CD19 or CD20-CAR T cells immunotherapy. Furthermore, we detected the factors affecting the efficacy and safety of therapy.
Methods
Literature searching and inclusion criteria
We searched the PubMed and EMBASE databases for relevant articles published up to September 5, 2016 with the search term “cart”. All studies related to the topics were included. All articles were published in English.
Literature screening
We extracted the data from each study: first author, year, number of patients, disease type, Ag recognition moieties, costimulatory domains, CART generation, original T cell sources (autologous or allogeneic), T cell culture time, transduction method, T cell treatment, CAR T cells persistence time, lymphodepletion, IL-2 infusion to patients, IL-2 infusion to cells, the infused total cell number, CAR T cells number, peak serum TNF level, peak serum IFN- γ level, peak serum IL-2 level, patients’ response to CAR T therapy, follow-up time and toxicity of the treatments.
There were two outcomes for the efficacy analysis. The primary efficacy outcome was patients’ response rate to CAR T therapy. Patients died not because of the disease or did not evaluate the response were not included for this analysis. The secondary efficacy outcome was patients’ progression free survival (PFS). For the safety analysis, we calculated the occurrence of toxicity of CAR T therapy and observed some frequent adverse events.
Statistic analysis
We used the Metaprop module in the R-3.3.2 statistical software package to analyze the response rate and the toxicity. Tests of heterogeneity were performed. When the I2 statistic was less than 50% and the p-value was more than 0.10, results were considered homogenous and a fixed-effect model was used. Otherwise, a random-effect model was used [18]. Subgroup analysis were performed to find the possible predictors.
We used Stata 12.0 to analyze PFS. All the factors analyzed in subgroup analysis of response were evaluated. PFS curves were assessed using the Kaplan–Meier method and compared by the log-rank test in the univariate meta-regression analysis. The independent prognostic factors of PFS was identified by cox regression model.
Contour-enhanced funnel plots was used to assess possible publication bias.
Results
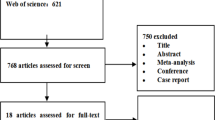
A total of 463 clinical trials were identified by the initial database search. A total of 18 articles were identified for analysis (Fig. 2).

Flow diagram of study selection process
Our study included 18 clinical trials and 185 B cell malignancies patients (126 leukemia and 59 lymphoma) received CAR T cells immunotherapy. The 126 leukemia patients included 39 chronic lymphocytic leukemia patients and 87 acute lymphocytic leukemia patients. The 59 lymphoma patients consisted of 31 diffuse large B-cell lymphoma, 11 mantle cell lymphoma, 7 non-Hodgkon’s lymphoma, 4 follicular lymphoma and 6 patients without detailed subtypes.
Treatment procedures
The characteristics of CAR T therapy were included in Table 1. Twelve patients in three trials were used with anti-CD20 CAR T. Three patients in one trial received third-generation CAR T with CD28, CD3ζ and CD137 (4-1BB) activation domains. OKT3, rHuIL-2, IL-15, LCL-irradiated, CD3/CD28 beads and CD19/CD80 artificial APCs were added into CAR T cells. The infused CAR T cell number ranged from 1.8 × 106 to 3.2 × 109.
Efficacy
Response rate
A total of 178 patients were eligible for the response rate evaluation. The overall response rate was 67% (95%CI: 53–79%) (Table 2). Subgroup analyses were performed, and the results were showed in Table 2. We observed that patients who received lymphodepletion had higher response rate (72%; 95%: 63–80%; P = 0.0405) than patients who did not (44%; 28–62%) (Additional file 1: Figure S1). Patients whose peak serum IL-2 level was over50 pg/mL had higher response rate (85%; 95%: 55–96; P = 0.04) than those less than50 pg/mL (31%; 95%: 6–74%) (Additional file 1: Figure S2). Results of other subgroup analyses were presented in Table 2.
Survival outcome
Progression free survival analysis included overall 90 patients from 15 clinical trials. The 6-month and 1-year PFS for this cohort were 65.62% (95%CL: 54.62–74.58%) and 44.18% (95%CL: 32.97–54.81%), respectively (Additional file 1: Figure S3A). The median and mean intervals of PFS were 10.4 and 21.62 (95%CL: 16.19–27.05) months, respectively. Association between patients’ PFS of CAR T cells immunotherapy and possible prognostic factors in univariate analysis were showed in Table 3. We observed that only CAR T cell costimulatory domains were related with PFS (p = 0.0489). The 1-year PFS of CD28 and CD3ζ (56.29%, 95%CL: 39.42–70.14%) was higher than that of CD137 and CD3ζ (33.39%, 95%CL: 16.56–51.22%) (Additional file 1: Figure S3B). The logrank test of other factors were showed in Table 3. Cox analysis showed that none factor was related to prognosis (Additional file 1: Table S1).
Safety
A total number of 154 patients were included in the overall analysis since two articles did not provide the data of the number of people with adverse events. The pooled estimate for overall incidence of any adverse events was 71% (95%CI: 0.49–0.92) (Additional file 1: Table S2). The estimate for incidence of grade ≥ 3 adverse events was 43% (95%CI: 0.23–0.63) within the related 154 patients (Additional file 1: Table S2).
After investigating grade ≥ 3 adverse events, we found that the most frequently occurred events included fatigue (18%, 95%CI: 0.12–0.24), night sweats (14%, 95%CI: 0.09–0.20), hypotension (12%, 95%CI: 0.08–0.19), injection site reaction (12%, 95%CI: 0.07–0.18), leukopenia (10%, 95%CI: 0.06–0.16), anemia (9%, 95%CI: 0.05–0.15) (Fig. 3).

Forest plot for most common adverse events and confidence internals
By subgroup analysis, we did not discover that serum IL-2, IFN-γ and TNF levels were correlated to the incidence of toxicities (Additional file 1: Table S2).
Publication bias
No potential publication bias was observed in funnel plot (Additional file 1: Figure S4).
Discussion
CAR T cells immunotherapy is rapidly developed in recent decades. How to improve the efficacy and reduce treatment toxicity remains the most concerned issues. Therefore, the following processions need to be improved: CAR design, gene transfection method, cytokine support, expansion and persistence of T cells, patients’ preconditioning, infusion dose of T-cells and types.
According to signaling domains, there were first, second and third generations of CAR. Data suggested that second-generation CARs with a costimulatory molecule mediated rapid activation, expansion, and persistence to T cells compared with first generation CARs, [19]. We discovered that the second-generation CAR T had a longer mean progression free survival time than first generation, but no significant difference(22.69 vs 18.52 months, P = 0.7754). Meanwhile, we didn’t find the difference in response (P = 0.7928) between first and second CARs. Therefore, the efficacy of second generation needs more research to verify. Because of the limited data, third generation was not evaluated. Whether third generation CARs are better than second generation CARs remains to be elucidated.
The costimulatory domain with second generation CAR T were usually used CD28 or CD137. Which domain shows better efficacy remains unknown. We discovered that no significant difference in the response rate between CD28 and CD137, but the CD137 signaling moieties in CARs related with lower survival (p = 0.0489). However, some studies exhibited that compared to CD28, the CD137 increased expansion and persistence of T cells [20, 21]. There were two possible reasons: first, CD137 was more novel, lacking of maturity; second, CD137-containing CARs could increase acute toxicity and the persistence of the infused T cells. There was no trial to compare the efficacy of costimulatory signal, therefore both basic and clinic trials are needed in this aspect.
CAR construction transducted to T cells by viral vector or electroporation. Viral transduction methods have higher transduction efficiency compared to electroporation, but it increases the risk of viral insertional oncogenesis. In our study, we did not find difference between the two methods. Considering only 5 patients transduced by electroporation, more trails are needed to detect gene transfer efficiency.
Should patients receive lymphodepletion or not, there was not been a common consensus by most researchers yet. Lymphodepletion regimen means depletion of recipient lymphocytes before CAR T cells infusion including chemotherapy, chemoradiotherapy, and monoclonal antibodies. It increased expansion, persistence, and efficacy of CAR T cells by eliminating regulatory T cells and other immune cells that may compete for cytokines, including IL-15 and IL-7, which activating antigen-presenting cell [22,23,24]. In this study, lymphodepletion was associated with better response (P = 0.0405), but no evidence of correlation with PFS, the same with the former article [15]. However, we didn’t perform subgroup analysis to assess the efficacy between different lymphodepletion regimens. In the future, research should focus on the effect of different lymphodepletion regimen on patients received CAR T cells.
Cytokine were often added to expanse T cells. Previous study presented that IL-2 promoted T-cell expansion to affect the efficacy [25]. We observed that peak serum IL-2 level in patients (P = 0.04) were positively associated with patients’ response to CAR T cells, in accordance with previous study. However, we observed that whether IL-2 administration to T cells or patients or not, the efficacy had no difference, not in accordance with former study [15]. These were two possible reasons for this result: first, the costimulatory domain could active antigen specific cytokine production cells without IL-2. Second, anti-CD3/anti-CD28 mAb-coated magnetic beads can stimulate T cell expansion without IL-2. Therefore, whether IL-2 administration to T cells or patients or not still needs more studies.
After infusion of CAR T cells, the cells will expanse to play a role and then go to apoptosis. Degree of expansion and duration of persistence is often considered to correlate with efficacy [26, 27]. However, we didn’t observe that expansion and persistence of T cells were related with efficacy. The following reasons should be considered for the result. First, previous study observed that costimulatory domain can increase persistence [28]. Next, other studies showed that lymphodepletion was benefitial to T-cell persistence and expansion in vivo [29, 30]. Meanwhile, IL-2 promoted T-cell expansion [25]. All these factors can influence efficacy. Consequently, during the process of CAR T therapy, more attention are needed to be paid in these procedures.
Commonly, the efficacy correlated with drug dose. There was no standard infusion dose of CAR T cells. Previous study defined the maximum tolerated CAR T cells dose as 1X 106 CAR T cells/ kg body weight [31]. The only existing reports failed to identify a correlation of transfused CAR T cells number and clinical efficacy. Also, the dose of administered CAR T cells could not predict peak blood levels of CAR T cells [12, 14] . These results were in accordance with our finding. We assume the reasons behind this may be that there were regulatory T cells repressed expansion in vivo. Meanwhile, interindividual variation may make significant differences.
Mature Th cells express the surface protein CD4 and are referred to as CD4+ T cells. They function in the activation of other immune cells by releasing T cell cytokines. Cytotoxic T cells killed virus-infected cells and tumor cells, and they are also related to transplant rejection. These cells are known as CD8+ T cells since they express the CD8 glycoprotein. Several studies observed that CD4+ and CD8+ contents and the proportion of T cells may affect efficacy [4, 32]. However, previous study reported that the absolute numbers of infused T-cell subsets did not appear to relate with clinical efficacy [4]. Our study didn’t analyze the proportion of CD4+/CD8+ whether related with efficacy with limited data. Further researches need be explored to find the optimal strategies.
Toxicity included CRS, on-target off-tumor effects and the toxicity caused by lymphodepletion. CRS can be caused by massive therapy-induced release of inflammatory cytokines. On-target off-tumor effects destroyed normal cells with the CAR-targeted antigens. We observed that the overall incidence of any adverse events was 71%, incidence of grade ≥ 3 adverse events was 43%, the most frequently occurred events included fatigue (18%), night sweats (14%), hypotension (12%), injection site reaction (12%) among the grade ≥ 3 adverse events. In patients after CAR T-cell infusion, IFN-γ and TNF are commonly high, which induces sepsis-like syndrome and causes organ failure [13]. However, these were not in accordance with our results. But we found that adverse events with higher IL-2, TNF, IFN-γ cytokine level happened more frequently. These factors were also closely related to CAR T-cell antitumor activity. Therefore, how to balance the efficacy and the toxicity should be further considered. A suicide gene, inducible caspase 9 (iCasp9) was integrated to CAR construction to regulate the persistence of CAR T-cells to control the on-target/off-tumor toxicities [33].
We included 18 articles to assess the efficacy and safety of CD19 or CD20-CAR T cells immunotherapy. Furthermore, we detected the factors affecting the efficacy and safety of therapy. However, our study has several limitations. First, the included articles were not totally prospective clinic studies, the potential performance bias might exist. Second, more studies were needed to assess the efficacy and sefety of CAR T therapy.
Conclusions
In conclusion, our study demonstrated a high response rate of CAR T therapy in refractory B cell malignancies. The study also showed lymphodepletion regimen and high serum IL-2 level were associated with better clinical responses, and that costimulatory domains was related with better PFS. Further modifications of CAR structure and optimal therapy strategy in continuing clinical trials are needed to obtain significant improvements.
Abbreviations
- AEs:
-
Adverse effects
- ALL:
-
Acute lymphocytic leukemia
- CAR T:
-
Chimeric antigen receptor- T cells
- CLL:
-
Chronic lymphocytic leukemia
- CM:
-
Costimulatory molecule
- CR:
-
Complete response
- CRS:
-
Cytokine release syndrome
- DLBCL:
-
Diffuse large B-cell lymphoma
- FL:
-
Follicular lymphoma
- ITAMs:
-
Immunoreceptor tyrosine-based activation motifs
- LCLs:
-
EBV-transformed lymphoblastoid B-cell lines
- MCL:
-
Mantle cell lymphoma
- MHC:
-
Major histocompatibility compleX
- NHL:
-
Non-Hodgkon’s lymphoma
- NR:
-
No response; NE: not evaluate
- PD:
-
Progress disease
- PR:
-
Partial response
- scFv:
-
Single-chain variable fragment
- SD:
-
Stable disease
- TAA:
-
Tumor associated antigen
- TCR:
-
T cell receptors
References
Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nature biotechnology. 2002;20(1):70–5.
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–84.
van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nature reviews Drug discovery. 2015;14(7):499-509.
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–28.
Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nature reviews Immunology. 2004;4(4):301–8.
Weijtens ME, Willemsen RA, Valerio D, Stam K, Bolhuis RL. Single chain Ig/gamma gene-redirected human T lymphocytes produce cytokines, specifically lyse tumor cells, and recycle lytic capacity. Journal of immunology (Baltimore, Md : 1950). 1996;157(2):836–43.
Wang X, Riviere I. Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer gene therapy. 2015;22(2):85–94.
Uckun FM, Jaszcz W, Ambrus JL, Fauci AS, Gajl-Peczalska K, Song CW, et al. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood. 1988;71(1):13–29.
Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101(4):1637–44.
Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;366(21):2008–16.
Jensen M, Tan G, Forman S, Wu AM, Raubitschek A. CD20 is a molecular target for scFvFc:zeta receptor redirected T cells: implications for cellular immunotherapy of CD20+ malignancy. Biol Blood Marrow Transplant. 1998;4(2):75–83.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33.
Zhang T, Cao L, Xie J, Shi N, Zhang Z, Luo Z, et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: a meta-analysis. Oncotarget. 2015;6(32):33961–71.
Xu XJ, Zhao HZ, Tang YM. Efficacy and safety of adoptive immunotherapy using anti-CD19 chimeric antigen receptor transduced T-cells: a systematic review of phase I clinical trials. Leuk Lymphoma. 2013;54(2):255–60.
Zhu Y, Tan Y, Ou R, Zhong Q, Zheng L, Du Y, et al. Anti-CD19 chimeric antigen receptor-modified T cells for B-cell malignancies: a systematic review of efficacy and safety in clinical trials. European journal of haematology. 2016;96(4):389–96.
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ (Clinical research ed). 2003;327(7414):557–60.
Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–74.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17(8):1453–64.
Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer research. 2011;71(13):4617–27.
Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. The Journal of experimental medicine. 2005;202(7):907–12.
Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–57.
Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends in immunology. 2005;26(2):111–7.
Caserta S, Alessi P, Basso V, Mondino A. IL-7 is superior to IL-2 for ex vivo expansion of tumour-specific CD4(+) T cells. European journal of immunology. 2010;40(2):470–9.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6.
Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–6.
Kohn DB, Dotti G, Brentjens R, Savoldo B, Jensen M, Cooper LJ, et al. CARs on track in the clinic. Molecular therapy : the journal of the American Society of Gene Therapy. 2011;19(3):432–8.
Cui Y, Zhang H, Meadors J, Poon R, Guimond M, Mackall CL. Harnessing the physiology of lymphopenia to support adoptive immunotherapy in lymphoreplete hosts. Blood. 2009;114(18):3831–40.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9.
Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Frontiers in pharmacology. 2014;5:235.
Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. 2010;16(9):1245–56.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. 2013;122(25):4129–39.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. 2013;5(177):177ra38.
Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. 2012;119(17):3940–50.
Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. 2013;122(17):2965–73.
Dai H, Zhang W, Li X, Han Q, Guo Y, Zhang Y, et al. Tolerance and efficacy of autologous or donor-derived T cells expressing CD19 chimeric antigen receptors in adult B-ALL with extramedullary leukemia. 2015;4(11):e1027469.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. 2014;6(224):224ra25.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. 2014;371(16):1507–17.
Wang Y, Zhang WY, Han QW, Liu Y, Dai HR, Guo YL, et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. 2014;155(2):160–75.
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. 2010;116(20):4099–102.
Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. 2016;127(24):2980–90.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Author information
Authors and Affiliations
Contributions
HZ collected, analyzed the data and wrote the article. XM provided the idea. Y.L. drew the figure. SZ modified the article. YL, XW, YZ, XO and TZ collected data, prepared the pictures and tables. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Forest plot for response rates and confidence internals in patients with or without lymphodepletion. Figure S2. Forest plot for response rates and confidence internals in patients with different serum IL-2 level. Figure S3. Progression-free survival (PFS) curves. A. the PFS for 90 patients; B. patients received CAR T cells with CD28 costimulatory domain had better PFS than CD137. Figure S4. funnel plot of substantial publication bias. Table S1. Cox regression analysis of patients’ PFS of CAR T cells immunotherapy and possible prognostic factors. Table S2. Subgroup analyses of adverse events. (DOCX 924 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, H., Luo, Y., Zhu, S. et al. The efficacy and safety of anti-CD19/CD20 chimeric antigen receptor- T cells immunotherapy in relapsed or refractory B-cell malignancies:a meta-analysis. BMC Cancer 18, 929 (2018). https://doi.org/10.1186/s12885-018-4817-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4817-4