
Abstract
Background
BK-UM (CRM197) is a mutant form of diphtheria toxin and a specific inhibitor of heparin-binding epidermal growth factor-like growth factor (HB-EGF). We assessed the safety, pharmacokinetics, recommended dose, and efficacy of BK-UM in patients with recurrent ovarian cancer (OC) or peritoneal cancer (PC), and measured HB-EGF levels in serum and abdominal fluid after BK-UM administration.
Methods
Eleven patients with advanced or recurrent OC or PC were enrolled and treated with BK-UM via the intraperitoneal route. The dose was escalated (1.0, 2.0, 3.3, and 5.0 mg/m2) using a 3 + 3 design.
Results
Eight of 11 patients completed treatment. No dose-limiting toxicity (DLT) was experienced at dose levels 1 (1.0 mg/m2) and 2 (2.0 mg/m2). Grade 3 transient hypotension as an adverse event (defined as a DLT in the present study) was observed in two of four patients at dose level 3 (3.3 mg/m2). Treatment with BK-UM was associated with decreases in HB-EGF levels in serum and abdominal fluid in seven of 11 patients and five of eight patients, respectively. Clinical outcomes included a partial response in one patient, stable disease in five patients, and progressive disease in five patients.
Conclusions
BK-UM was well tolerated at doses of 1.0 and 2.0 mg/m2, with evidence for clinical efficacy in patients with recurrent OC or PC. A dose of 2.0 mg/m2 BK-UM is recommended for subsequent clinical trials.
Trial registration
This trial was prospectively performed as an investigator-initiated clinical trial. The trial numbers are UMIN000001002 and UMIN000001001, with registration dates of 1/30/2008 and 2/4/2008, respectively. UMIN000001001 was registered as a trial for the continuous administration of BK-UM after UMIN000001002.
Similar content being viewed by others


Background
Ovarian cancer (OC) is a lethal gynecologic malignancy that has an extremely poor prognosis, due to the extension of OC cells into the peritoneal cavity [1]. Metastasis from epithelial ovarian cancer (EOC) can occur via hematogenous, lymphatic, or transcoelomic routes, of which the last is the most common, and is responsible for the highest prevalence of morbidity and mortality in patients with OC [1]. Most patients with recurrent OC experience massive ascites and ileus associated with transcoelomic metastasis, and preventing transcoelomic metastasis may thus play a key role in improving the prognosis in OC patients.
In the past decade, cytoreductive surgery and the introduction of platinum plus taxane-based chemotherapies have improved survival in patients with EOC [2, 3]. In the International Collaborative Ovarian Neoplasm 7 trial, standard chemotherapy with bevacizumab failed to increase overall survival in patients with newly diagnosed OC, but did improve progression-free survival and retention of peritoneal fluid [4]. Platinum is recognized as a key drug for OC therapy [5], and the development of biological agents and molecular-targeted therapeutics with low toxicity are needed to overcome resistance or restore sensitivity to platinum.
Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is an epidermal growth factor receptor (EGFR) ligand [6]. HB-EGF is initially synthesized as a transmembrane protein (proHB-EGF), which is then cleaved by a protease at the cell surface to yield the mitogenic-active soluble form of HB-EGF (sHB-EGF) via ectodomain shedding [7]. sHB-EGF is a potent mitogen that promotes cell adhesion, cell motility, and angiogenesis [8]. HB-EGF also contributes to resistance to chemotherapy [9]. HB-EGF expression was shown to be enhanced in cancer tissues or peritoneal tissues in OC patients [10–13], and high expression of HB-EGF in cancer tissues was significantly associated with a poor prognosis in OC [10]. Emerging evidence suggests that HB-EGF is a rational therapeutic target for cancers, including OC, gastric cancer, and breast cancer [14, 15].
CRM197 is the active ingredient of the investigational drug BK-UM. CRM197 was originally isolated as a non-toxic mutant form of diphtheria toxin (DT), and has since been shown to inhibit the proliferation of cancer cells by three modes of action [16–18]. CRM197 strongly inhibited tumor growth in nude mice in various cell systems [10]. However, CRM197 displays 106-fold less toxicity than DT [17], which is recognized as being extremely toxic, Massive release of DT is likely to cause lethal necrosis in the heart and liver, with a reported lethal dose in humans of about 0.1 μg/kg body weight [19]. It is therefore necessary to monitor any potential severe adverse effects of BK-UM, including cardio-, neuro-, or nephrotoxicities, irrespective of its route of administration.
CRM197 was safe and relatively well tolerated when injected subcutaneously via the abdominal wall on alternate days for 6 days at 1.7, 2.6, and 3.5 mg/day in patients with recurrent cancer [20]. In a pre-clinical study, we reported that daily intraperitoneal administration of BK-UM, which has demonstrated a potentially dose-dependent anti-cancer effect, may be the optimal regimen [21]. Herein, we report the results of a phase-I study designed to assess the safety, pharmacokinetics, recommended phase-II dose, and efficacy of BK-UM in patients with recurrent OC or peritoneal cancer (PC).
Methods
Patient selection
This clinical trial was undertaken as an open-label, phase-I dose-escalation study, and was performed as a prospective investigator-initiated clinical trial. The study was conducted at Fukuoka University Hospital (Fukuoka, Japan) in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization’s Good Clinical Practice Guidelines. The study protocol was approved by the Institutional Review Board of Fukuoka University Hospital.
Study-inclusion criteria were:(i) EOC or primary PC confirmed by histology; (ii) recurrent OC or PC within 6 months after completion of a primary treatment containing platinum and a taxane (including patients who had received second- or subsequent-line chemotherapy after recurrence following primary treatment containing platinum and a taxane); (iii) performance status 0 or 1 according to the Eastern Cooperative Oncology Group classification; (iv) evidence of adequate liver, kidney, and bone marrow functions (absolute white blood cell count >3000/μL, or neutrophil count ≥1000/μL, platelet count ≥100,000/μL, hemoglobin ≥8.0 g/dL, aspartate aminotransferase ≤2.5-fold the upper limit of normal, alanine transaminase ≤2.5-fold the upper limit of normal, serum total bilirubin ≤1.5 mg/dL, serum creatinine ≤1.5 mg/mL); (v) detectable levels of HB-EGF in serum or peritoneal fluid; (vi) diphtheria antibody titer <0.1 IU/mL; and (vii) practicality of placing a port in the abdomen.
All subjects underwent electrocardiography to confirm the absence of abnormalities requiring treatment, and sensory and motor nerve-conduction studies to evaluate peripheral neuropathy.
Exclusion criteria were: (i) history of cardiac disease ≥ III according to the New York State Heart Association classification; (ii) <50% left ventricular ejection fraction by echocardiography; (iii) myocardial infarction within 6 months of study enrollment; (iv) uncompensated liver cirrhosis; (v) severe lung disease necessitating oxygen inhalation; (vi) psychotic disorders; (vii) uncontrolled diabetes mellitus; (viii) ileus or subileus; (ix) uncontrolled infectious disease; (x) peripheral neuropathy of grade ≥2; (xi) pregnancy or breastfeeding; (xii) life expectancy <3 months; (xiii) receipt of an investigational agent within 28 days before study enrollment; and (xiv) history of administration of antiserum.
Concurrent investigational or antineoplastic agents were not permitted during the study.
Investigational drug
CRM197 was produced from Corynebacterium diphtheria C7β (197) and purified by the Kanonji Institute at the Research Foundation for Microbial Diseases of Osaka University (Osaka, Japan). BK-UM contained CRM197, sodium phosphate, and sucrose. BK-UM was also produced at the above institute according to good manufacturing practice.
Study design
This was a dose-escalation study with a 3 + 3 trial design at each dose to determine the maximum-tolerated dose (MTD) and recommended dose. BK-UM was administered intraperitoneally at doses of 1.0, 2.0, 3.3, and 5.0 mg/m2, 5 days a week for 2 weeks, based on the results of a pre-clinical study [21]. An anti-histamine was administered as premedication before BK-UM injection. Dose escalation of BK-UM was investigated by the Data and Safety Monitoring Committee (DSMC). Treatment of the first patient was finished completely, and the safety of BK-UM was then reviewed by the DSMC. If no dose-limiting toxicity (DLT) was observed in three patients at a given dose, the next three patients were treated with the next-higher dose. If DLT was experienced by one or two of the three patients at any dose, three additional patients were treated with the same dose. If two or fewer of six patients treated with a given dose exhibited DLT, this dose was considered to exceed the MTD and dose escalation was halted. The recommended phase-II dose was defined as one dose level lower than the MTD. Patients who progressed after showing a response to BK-UM were not allowed to be re-treated with BK-UM. If more than two patients withdrew from the present study, safety was evaluated by the DSMC. Conventional chemotherapy was administered in patients who requested it after the observation period of the trial, for as long as there was a diagnosis of progressive disease, or for six courses in patients with stable disease (SD), partial response (PR), or complete response.
Safety evaluation and definition of DLT
Safety was evaluated every week during treatment and until 6 weeks after the first infusion of BK-UM. Safety of BK-UM was evaluated according to adverse events (AEs) detected via vital signs, physical signs, whole blood cell counts, serum chemistry, urinalyses, 12-lead electrocardiography, cardiac monitoring, echocardiography, muscle tests, neural reflex tests, nerve conduction velocity testing, and thoracoabdominal radiographs. Laboratory tests for in cardiotoxicity were also undertaken.
All AEs were graded according to the Common Terminology Criteria for Adverse Events v3.0 set by the National Cancer Institute. DLT was defined as an AE or laboratory abnormality that occurred 6 weeks after the first infusion of BK-UM. A DLT was judged to be related to BK-UM if it met any of the following criteria: hematologic toxicity grade ≥4; gastrointestinal disorder possibly due to peritonitis carcinomatosa grade ≥4; or non-hematologic toxicity grade ≥3. Safety data were evaluated by the DSMC at all doses.
Efficacy assessments
Responses were evaluated 6 weeks after the first infusion of BK-UM. Recurrent lesions were evaluated by computed tomography before infusion of BK-UM, to provide baseline images. Clinical evaluation of response was estimated using baseline computed tomography images and findings 6 weeks after the first infusion of BK-UM, according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.0 guidelines. The interval for overall survival was defined as the period from the day of the first infusion of BK-UM to the day on which death occurred. Some patients were discharged from Fukuoka University Hospital after the end of this clinical trial and treated at other hospitals, and the dates of death for these patients were recorded using information acquired from those hospitals.
Pharmacokinetics
Blood samples were collected before and at 1, 3, 6, and 12 h after infusion of each dose on day 1. Pre-dose samples were collected on days 2–5 and 8–12. Samples were collected in a heparinized tube and centrifuged at 3000 × g for 10 min at room temperature. Plasma samples were stored at −80 °C for analysis of BK-UM concentrations. Plasma concentrations of BK-UM were determined by enzyme-linked immunosorbent assay (ELISA). Plates were coated with an anti-DT monoclonal antibody to capture CRM197, followed by the addition of diluted plasma samples obtained from patients, and a horseradish peroxidase-labeled monoclonal antibody against DT. The sensitivity of this assay was ≥20 ng/mL.
Measurement of HB-EGF levels in serum and abdominal fluid
Blood and abdominal fluid were withdrawn from all patients into anticoagulant-excluded tubes before and after infusion of BK-UM. HB-EGF concentrations in serum and abdominal fluid were determined by a modified enzyme assay [22] using a sandwich ELISA (DuoSet; R&D Systems, Minneapolis, MN, USA).
Results
Patient characteristics
This trial was conducted at 12/10/2007. Eleven of 50 patients with recurrent cancer were enrolled, including 10 with OC and one with primary PC (Table 1). The remaining 39 patients were not eligible for the following reasons: 12 patients had diphtheria antibody levels >0.1 IU/mL; 21 and five patients had performance statuses of 2 or 3, respectively; and one patient recurred more than 6 months after primary treatment containing platinum and a taxane. Among the patients who received BK-UM at 1.0 mg/m2 (level 1 dose), one patient had leptomeningeal disease previously treated using a γ-knife, but had been in a stable condition for 4 months. Four patients (numbers 1–4) were enrolled at 1.0 mg/m2 BK-UM (dose level 1) because one patient who lived in Tokyo wanted to return to Tokyo, and withdrew their consent soon after BK-UM treatment.
Eight patients completed the study according to the protocol, one patient withdrew their consent as noted above, and two patients dropped out of the trial because of DLTs. All 11 enrolled patients were evaluated for safety and efficacy on an intent-to-treat basis.
Safety and tolerability
All AEs of grade ≥2 are listed in Table 2. All patients experienced at least one AE. At BK-UM doses of 1.0 and 2.0 mg/m2 (dose levels 1 and 2), all AEs were considered unlikely to have a causal association with BK-UM. Grade 3 AEs such as ileus, abdominal fullness, and anemia were observed in one patient at 2.0 mg/m2 BK-UM (dose level 2). Most patients had grade 1 fever between days 1 and 3 of BK-UM treatment, but neither the frequency nor severity of these AEs increased with dose escalation. BK-UM was thus generally considered to be well-tolerated at doses of 1.0 and 2.0 mg/m2. However, both patients treated at 3.0 mg/m2 BK-UM (dose level 3) developed grade 3 hypotension on day 8 of treatment, which was defined as a DLT. This AE manifested as nausea, abdominal pain, and transient hypotension, accompanied by irritation of the peritoneum, fever, and elevated C-reactive protein levels. Complete recovery from all AEs was observed within 1 week in both patients. All other AEs observed at dose level 3 were grade ≤2. No severe AEs (grades 4 or 5), including severe cardiotoxicity- or neurotoxicity-related DLTs, were found in any patient at any dose level.
Dose escalation and MTD
No DLT was observed at dose levels 1 and 2, but two patients at dose level 3 showed grade 3 hypotension 8 days after the first infusion. The MTD was therefore judged to be at dose level 3 (3.0 mg/m2), and the recommended dose was determined to be 2.0 mg/m2.
Pharmacokinetics
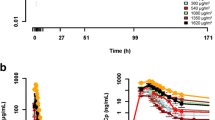
The plasma concentration of BK-UM after intraperitoneal administration was measured 15 times per course in each patient. At 1.0 mg/m2 BK-UM (dose level 1), plasma BK-UM was only detected in three of 60 samples from four patients. BK-UM plasma levels were detected in nine of 45 samples from three patients at 2.0 mg/m2 (level 2) and 11 of 51 samples from four patients at 3.0 mg/m2 (level 3). The plasma concentration of BK-UM was unmeasurable throughout the treatment with BK-UM in five patients, while BK-UM was only sporadically detected in plasma in the remaining six patients. These results suggested that intraperitoneal delivery of BK-UM did not result in clinically relevant plasma exposure (Additional file 1: Table S1).
Evaluation of response
BK-UM treatment resulted in reductions in HB-EGF serum concentrations in five patients, in abdominal fluid in four patients, and in abdominal fluid and serum in four patients (Table 3). Patients 2, 8, and 11 were not included in this analysis because HB-EGF concentrations were not measured on the scheduled day after BK-UM treatment in these patients. The present results suggested that BK-UM reduced HB-EGF concentrations in abdominal fluid or serum if the HB-EGF concentration was ≥125 pg/mL. Serum levels of cancer antigen-125 also decreased in two of 11 patients. DT antibody levels were increased in all patients after BK-UM treatment. One of the 11 patients achieved PR (Table 3), five patients showed SD, and the remaining five patients showed progressive disease. In addition to the abdominal cavity, patients 4, 6, and 9 had metastatic lesions in the brain, liver, and lungs, respectively (though patient number 4 had a history of leptomeningeal disease previously treated by γ-knife, and had been in a stable condition for 4 months). The metastatic lesions in the abdominal cavity in these three patients were markedly reduced after BK-UM treatment, even though the extra-abdominal lesions had progressed or were stable. At least three patients received intraperitoneal administration of BK-UM 1 mg/m2 or 2 mg/m2 for 2 weeks. Only patients 1 and 4 (level 1) received two courses of intraperitoneal BK-UM (1 mg/m2), and responses in these patients were evaluated at week 8 after the start of the first course of BK-UM, compared with evaluation at week 6 in the other patients who only received one course of intraperitoneal BK-UM. The benefit of intraperitoneal administration of BK-UM thus lasted up to 6 weeks in patients 3, 5, 10, and 11, and 8 weeks in patients 1 and 4. Conventional chemotherapy was started at week 9 in patients 1 and 4, and week 7 in patients 3, 7, 8, 10, and 11. Patients 2, 5, 6, and 9 requested no further therapy. Five of the seven patients who received further conventional chemotherapy exhibited overall survival beyond 14 months (Table 4). Three patients (1, 3, and 11) showed no evidence of disease following BK-UM treatment and subsequent therapy, and one patient (11) remained free of disease.
Discussion
HB-EGF is a ligand that binds to the EGFR. Although several studies have investigated anti-cancer agents targeting the EGFR and other ERBB family receptors, few clinical trials have investigated the use of agents targeting EGF family growth factor ligands [23]. Previous studies have suggested that HB-EGF is expressed at high levels in OC patients, and represents a promising target for OC treatment [10, 14]. However, the current study provides the first evidence of HB-EGF values in OC patients.
A previous phase-I study of intraoperative intraperitoneal administration of 60–100 mg/m2 cisplatin detected a maximum plasma concentration of cisplatin of 0.7–2.3 μg/mL after 1–1.5 h, giving a mean peritoneal-to-plasma area-under-the-curve ratio during perfusion of 19.5 for 100 mg/m2 [24]. The plasma cisplatin level ranged from one-third to one-tenth of the total amount of intraperitoneally administered cisplatin. Conversely, the plasma BK-UM level was >20 ng/mL. Intraperitoneal administration of 1–3.3 mg/m2 BK-UM would be expected to result in plasma levels of ≥100 ng/mL; however, BK-UM was undetectable in most plasma samples in the current study. There are several possible reasons for this phenomenon: (i) BK-UM may not pass through peritoneal membranes readily because of its high molecular weight; (ii) BK-UM may be absorbed readily into the lymphatic system in the peritoneum; and (iii) most BK-UM may bind rapidly to the peritoneum, with high expression of HB-EGF on peritoneal membranes. In the present study, even patients with DLTs had undetectable plasma levels of BK-UM, suggesting that AEs are induced by local administration of BK-UM.
Hypersensitivity was mitigated by administration of an anti-histamine in all patients before intraperitoneal administration of BK-UM. Although fever (a common AE) occurred up to 3 days after the beginning of BK-UM administration, it was not accompanied by general fatigue, suggesting that the fever was generated locally. Other AEs, including nausea, vomiting, appetite loss, and abdominal fullness, could have been related to recurrent disease. Two patients treated at dose level 3 developed transient hypotension as a DLT at day 8, associated with uncontrolled abdominal pain and elevated C-reactive protein levels. Two patients with DLTs at dose level 3 did not have ascites, whereas the other two patients who did have ascites had no severe AEs. Transient hypotension could have been mediated by a parasympathetic reaction related to stimulation of the peritoneum by BK-UM, which could have caused inflammation in the abdominal cavity. It was also notable that both patients developed DLT hypotension at day 8. BK-UM suppresses the function of HB-EGF in the peritoneum, suggesting that it may be associated with blocking the circulation of peritoneal fluid, and high concentrations of BK-UM may thus cause excessive peritoneal stimulation. In a phase-I study of a monoclonal antibody (KHK2866) against HB-EGF, neurotoxicity manifested as complex partial seizures, aphasia, and confusion after first-dose intravenous administration, but these AEs were reversible and unpredictable [23, 25]. In the present study, muscle tests, neural reflex tests, and nerve conduction velocity tests revealed no neurotoxic abnormalities in any patient. However, in the event of intravenous administration of BK-UM, patients should be closely monitored for severe AEs such as cardiotoxicity, neurotoxicity, and nephrotoxicity.
The present study enrolled patients who had progressed to advanced disease despite previous treatments with several chemotherapeutic regimens. In addition, safety evaluations were performed 8 weeks after enrollment in patients who received one cycle of the present study treatment. Chemotherapeutic regimens are usually repeated for 3–4 weeks in OC patients, and demonstrating clinically favorable efficacy of BK-UM would thus be difficult because of the long observation period in the current study. However, one of five patients achieved PR or SD, suggesting that BK-UM may be effective against peritoneal dissemination of OC. Seven of 11 patients were treated with conventional chemotherapy after intraperitoneal administration of BK-UM, five of whom survived for >14 months. These five patients also had PR or SD, and four of them received another platinum-based chemotherapeutic regimen. In principle, the prognosis of OC depends on the tumor’s sensitivity to platinum [5], and patients who are refractory or resistant to platinum treatment showed a sensitivity of <10% to second-line chemotherapy [5]. These results suggest that BK-UM may represent a promising anti-cancer agent for OC therapy, by rescuing platinum sensitivity by improving peritoneal dissemination. Conversely, intrahepatic lesions or pleural effusions were exacerbated by intraperitoneal administration of BK-UM, though it is possible that replacing local intraperitoneal therapy with systemic intravenous administration of BK-UM could improve intrahepatic lesions or pleural effusions.
HB-EGF levels in serum have been shown to be correlated with levels in abdominal fluid and cancer tissues in OC patients [22]. However, HB-EGF is cleaved by a disintegrin and metalloproteinase (ADAM) and other membrane proteases, including membrane-type 1 matrix metalloprotease (MT1-MMP) [26]. However, the form of HB-EGF measured by ELISA in the current study was only cleaved by ADAM (preliminary results), while the form of HB-EGF cleaved by ADAM and MT-MMPs can be blocked by BK-UM [26]. In our study, BK-UM treatment elicited SD in some patients with low HB-EGF levels. Levels of the active form of HB-EGF might be elevated in patients with recurrent OC. The development of a kit for measuring all forms of HB-EGF is needed to facilitate further studies of BK-UM.
Conclusion
The results of this first-in-human study demonstrated that BK-UM is safe and well tolerated for the treatment of OC, with an MTD and recommended dose for phase-II studies of 3.3 mg/m2 and 2.0 mg/m2, respectively. Further investigations should focus on the: (i) route of administration of BK-UM; (ii) the development of a kit for measuring associated targeted molecules; and (iii) the combination of conventional anti-cancer agents with BK-UM.
Abbreviations
- ADAM:
-
A disintegrin and metalloproteinase
- AE:
-
Adverse event
- DLT:
-
Dose-limiting toxicity
- DT:
-
Diphtheria toxin
- EGFR:
-
Epidermal growth factor receptor
- ELISA:
-
Enzyme-linked immunosorbent assay
- EOC:
-
Epithelial ovarian cancer
- HB-EGF:
-
Heparin-binding epidermal growth factor-like growth factor
- MTD:
-
Maximum-tolerated dose
- MT-MMP:
-
Membrane-type matrix metalloprotease
- OC:
-
Ovarian cancer
- PC:
-
Peritoneal cancer
- PR:
-
Partial response
- SD:
-
Stable disease
- sHB-EGF:
-
Soluble form of heparin-binding epidermal growth factor-like growth factor
References
Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006;7:925–34.
Crawford SC, Vasey PA, Paul J, Hay A, Davis JA, Kaye SB. Does aggressive surgery only benefit patients with less advanced ovarian cancer? Results from an international comparison within the SCOTROC-1 Trial. J Clin Oncol. 2005;23:8802–11.
Trimble EL, Wright J, Christan MC. Treatment of platinum-resistant ovarian cancer. Expert Opin Pharmacother. 2001;2:1299–306.
Oza AM, Cook AD, Pfisterer J, Embleton A, Ledermann JA, Pujade-Lauraine E, et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015;16:928–36.
Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376–88.
Massagué J, Pandiella A. Membrane-anchored growth factors. Annu Rev Biochem. 1993;62:515–41.
Goishi K, Higashiyama S, Klagsbrun M, Nakano N, Umata T, Ishikawa M, et al. Phorbol ester induces the rapid processing of cell surface heparin-binding EGF-like growth factor: conversion from juxtacrine to paracrine growth factor activity. Mol Biol Cell. 1995;6:967–80.
Raab G, Klagsbrun M. Heparin-binding EGF-like growth factor. Biochim Biophys Acta. 1997;1333:F179–99.
Wang F, Liu R, Lee SW, Sloss CM, Couget J, Cusack JC. Heparin-binding EGF-like growth factor is an early response gene to chemotherapy and contributes to chemotherapy resistance. Oncogene. 2007;26:2006–16.
Miyamoto S, Hirata M, Yamazaki A, Kageyama T, Hasuwa H, Mizushima H, et al. Heparin-binding EGF-like growth factor is a promising target for the therapy of ovarian cancer. Cancer Res. 2004;64:5720–7.
Tanaka Y, Miyamoto S, Yagi H, Sonoda K, Kobayashi H, Kishikawa T, et al. Clinical significance of heparin-binding epidermal growth factor-like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer. Clin Cancer Res. 2005;11:4783–92.
Yagi H, Miyamoto S, Tanaka Y, Sonoda K, Kobayashi H, Kishikawa T, et al. Clinical significance of heparin-binding epidermal growth factor-like growth factor in peritoneal fluid of ovarian cancer. Br J Cancer. 2005;92:1737–45.
Yagi H, Yotsumoto F, Miyamoto S. Heparin-binding epidermal growth factor-like growth factor promotes transcoelomic metastasis in ovarian cancer through epithelial-mesenchymal transition. Mol Cancer Ther. 2008;7:3441–51.
Miyamoto S, Yagi H, Yotsumoto F, Kawarabayashi T, Mekada E. Heparin-binding epidermal growth factor-like growth factor as a novel targeting molecule for cancer therapy. Cancer Sci. 2006;97:341–7.
Yotsumoto F, Yagi H, Suzuki SO, Oki E, Tsujioka H, Hachisuga T, et al. Validation of HB-EGF and amphiregulin as targets for human cancer therapy. Biochem Biophy Res Commun. 2008;365:555–61.
Mitamura T, Higashiyama S, Taniguchi N, Klagsbrun M, Mekada E. Diphtheria toxin binds to the epidermal growth factor (EGF)-like domain of human heparin-binding EGF-like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J Biol Chem. 1995;270:1015–9.
Kageyama T, Ohishi M, Miyamoto S, Mizushima H, Iwamoto R, Mekada E. Diphtheria toxin mutant CRM197 possesses weak EF2-ADP-ribosyl activity that potentiates its anti-tumorigenic activity. J Biochem. 2007;142:95–104.
Hamaoka M, Chinen I, Murata T, Takashima S, Iwamoto R, Mekada E. Anti-human HB-EGF monoclonal antibodies inhibiting ectodomain shedding of HB-EGF and diphtheria toxin binding. J Biochem. 2010;148:55–69.
Pappenheimer Jr AM. Diphtheria toxin. Annu Rev Biochem. 1977;46:69–94.
Buzzi S, Rubboli D, Buzzi G, Buzzi AM, Morisi C. CRM197 (nontoxic diphtheria toxin): effects on advanced cancer patients. Cancer Immunol Immunother. 2004;53:1041–8.
Nam SO, Yotsumoto F, Miyata K, Suzaki Y, Yagi H, Odawara T, et al. Pre-clinical study of BK-UM, a novel inhibitor of HB-EGF, for ovarian cancer therapy. Anticancer Res. 2014;34:4615–20.
Hikita S, Yotsumoto F, Fukami T, Horiuchi S, Sanui A, Miyata K, et al. Assessment of HB-EGF levels in peritoneal fluid and serum of ovarian cancer patients using ELISA. Anticancer Res. 2011;31:2553–9.
Sarantopoulos J, Mita MM, Birrer MJ, Cranmer LD, Campos LT, Zhang X, et al. Phase 1 study of monotherapy with khk2866, an anti-heparin-binding epidermal growth factor-like growth factor monoclonal antibody, in patients with advanced cancer. Target Oncol. 2016;11:317–27.
Zivanovic O, Abramian A, Kullmann M, Fuhrmann C, Coch C, Hoeller T, et al. HIPEC ROC I: a phase I study of cisplatin administered as hyperthermic intraoperative intraperitoneal chemoperfusion followed by postoperative intravenous platinum-based chemotherapy in patients with platinum-sensitive recurrent epithelial ovarian cancer. Int J Cancer. 2015;136:699–708.
Kasai N, Adachi M, Yamano K. Preclinical pharmacokinetics evaluation of anti-heparin-binding EGF-like growth factor (HB-EGF) monoclonal antibody using cynomolgus monkeys via 89Zr-immuno-PET study and the determination of drug concentrations in serum and cerebrospinal fluid. Pharm Res. 2016;33:476–86.
Koshikawa N, Mizushima H, Minegishi T, Eguchi F, Yotsumoto F, Nabeshima K, et al. Proteolytic activation of heparin-binding EGF-like growth factor by membrane-type matrix metalloproteinase-1 in ovarian carcinoma cells. Cancer Sci. 2011;102:111–6.
Acknowledgements
Not applicable.
Funding
This study was supported by a Grant-in-Aid for Research Promotion for Innovative Therapies against Cancer Program and Translational Research Network Program from the Ministry of Education, Culture, Sports, Science and Technology (Tokyo, Japan) to E. Mekada; a Grant-in-Aid for Scientific Research (B) (No. 26293362), a Grant-in-Aid for Challenging Exploratory Research (No. 26670731), the Center for Advanced Molecular Medicine, Fukuoka University from the Ministry of Education, Culture, Sports, Science and Technology (Tokyo, Japan), and a Grant-in-Aid from the Kakihara Science and Technology Foundation (Fukuoka, Japan) to S. Miyamoto.
The roles of each funding agency were as follows: Research Promotion for Innovative Therapies against Cancer Program and Translational Research Network Program from the Ministry of Education, Culture, Sports, Science and Technology (Tokyo, Japan) for collection, analysis and interpretation of data; Scientific Research (B) (No. 26293362) for study design and writing of the report; Challenging Exploratory Research (No. 26670731) for study design and writing of the report; Center for Advanced Molecular Medicine, Fukuoka University from the Ministry of Education, Culture, Sports, Science and Technology (Tokyo, Japan) for study design; Kakihara Science and Technology Foundation (Fukuoka, Japan) for writing of the report.
Availability of data and materials
Any additional supporting data involving details of clinical data can be found in the electronic medical record system of the first affiliated hospital of Fukuoka University and are available on request from the corresponding author.
Authors’ contributions
Conception and design of the study: SM (Shingo Miyamoto), EM Acquisition of data: SM (Shingo Miyamoto), FY, TU, TF, AS, KM, SON, SF, TK, MM, HK, DM, KS (Kyoko Shirota), TY, MK, HN, KS (Keijiro Saku), YT, KI, YT, KT, AM, TH, SN, TO, KM, SM (Sadao Manabe), TI, YO, MO, TH, HM, RI, EM Analysis and interpretation of data: SM (Shingo Miyamoto), FY, TU, TF, AS, KM, SON, SF, TK, MM, HK, DM, KS (Kyoko Shirota), TY, MK, HN, KS (Keijiro Saku), YT, KI, YT, KT, AM, TH, SN, TO, KM, SM (Sadao Manabe), TI, YO, MO, TH, HM, RI, EM Drafting of the article: SM (Shingo Miyamoto), EM Critical revision of the article for important intellectual content: SM (Shingo Miyamoto), EM Final approval of the article: SM (Shingo Miyamoto). All authors have read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All patients provided written informed consent for the publication of their individual details.
Ethics approval and consent to participate
This clinical trial was undertaken as an open-label, phase-I dose-escalation study, conducted at Fukuoka University Hospital (Fukuoka, Japan). It was conducted in accordance with the principles of the Declaration of Helsinki and International Conference on Harmonization’s Good Clinical Practice Guidelines. The study protocol was approved by the Institutional Review Board of Fukuoka University Hospital. All patients provided written informed consent to participate in this study.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Table S1. Pharmacokinetics of BK-UM. (PDF 48 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Miyamoto, S., Yotsumoto, F., Ueda, T. et al. BK-UM in patients with recurrent ovarian cancer or peritoneal cancer: a first-in-human phase-I study. BMC Cancer 17, 89 (2017). https://doi.org/10.1186/s12885-017-3071-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-017-3071-5