
Abstract
Background
Primary natural killer (NK)/T cell lymphoma of adrenal glands is an extremely rare tumor with aggressive clinical behavior. There have only been a few cases reported worldwide and the highest reported survival was 90 days. We report the first case of primary unilateral adrenal NK/T cell lymphoma in China with good outcome.
Case presentation
This is a 28-year-old man who presented with abdominal pain and was found to have a large mass on the left adrenal and the top of the renal region. The patient underwent surgical resection and the pathology revealed primary adrenal NK/T cell lymphoma. He received adjuvant sandwich therapy encompassing sequential chemotherapy, radiotherapy and chemotherapy. The patient remains clinically and symptomatically disease-free with over two years follow up.
Conclusion
Given the rarity of this disease, there is limited experience with regard to its diagnosis and treatment. This case report will add to the scant literature on this tumor and will be useful for the differential diagnosis and treatment of adrenal disease.
Similar content being viewed by others

Background
There have been seven reported cases of NK/T cell lymphoma involving adrenal masses worldwide [1–7]. Among them, only one was primary adrenal NK/T cell lymphoma [2]. There have been no reported cases of the primary adrenal NK/T-cell lymphoma in China. Anatomically, it usually originates in the nasal cavity/nasopharynx and invades the surrounding tissues. Sometimes, it can arise in other organs including the skin, spleen/liver, or the gastrointestinal tract [8]. Here, we report a primary unilateral adrenal NK/T cell lymphoma with subsequent management. Although the longest survival was reported to be 90 days for adrenal NK/T cell lymphoma, our patient has been disease-free for 26 months.
Case presentation
Case report
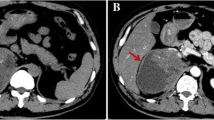
A 28-year-old male experienced abdominal pain and a mass on the left side of the abdomen. Color duplex ultrasonogram and subsequent computed tomography (CT) scan revealed a 10 × 9 cm adrenal mass invading the top of left renal and partial of retroperitoneal region (Fig. 1. a and b). He underwent surgical removal of the mass in an outside hospital. The pathology specimens were evaluated in our hospital which showed neoplastic infiltration of pleomorphic lymphoid cells with scanty cytoplasm, irregular nuclear contour, and prominent nucleoli. Immunohistochemistry analysis showed that the tumor cells were positive for CD2, Bcl-2, CD43, CD56, negative for CD20, CD4, CD5, CD7, CD8, CD138, PCK, HMB45, Des, Pax-5, Bcl-6, Vim, CD34, TdT, MPO and equivocal for CD3. Ki67 labelling index was 40% (Fig. 2). Chromogenic in situ hybridization (CISH) was positive for the EBER gene (Fig. 3). These are consistent with NK/T-cell lymphoma.

Abdominal CT scans. An abdominal axial CT scan indicates a large left adrenal mass that had invaded retroperitoneal cavity before surgery (a). A reconstructed coronal CT image shows that the top of left kidney was invaded by the tumor (b). An abdominal axial CT scan shows a residual mass after the surgery (c). An abdominal axial CT scan shows a complete response after the postoperative chemotherapy with four cycles of CHOP (d)

Histological and immunohistochemical analysis. Neoplastic infiltrate of relatively pleomorphic lymphoid cells with scanty cytoplasm, irregular nuclear contour, and prominent nucleoli (hematoxylin and eosin, magnification × 400). Immunohistochemical staining demonstrates positive (magnification × 400) for CD43, CD2, CD56, but negative (magnification × 200) for CD20, CD5, CD7, Pax-5

The expression of EBER. Chromogenic in situ hybridization (CISH) was positive for the EBER gene. (magnification × 400)
After the surgery, the patient was without any systemic symptoms. He did have a 10 kg weight loss secondary to the removal of the majority of the large mass and the poor postoperative nutrition support. The postoperative physical examination was unremarkable. A postoperative CT scan of the abdomen showed a residual tumor, measuring 2.7 × 5.3 cm at the left side of retroperitoneum with the absence of left adrenal gland and kidney (Fig. 1. c). The bone marrow biopsy was negative. Nasal magnetic resonance imaging (MRI) showed normal findings. The contrast CT scans of the neck, chest and pelvis had been done and showed normal tissue structure. Primary unilateral adrenal NK/T cell lymphoma was diagnosed. He received four cycles of chemotherapy with cyclophophamide, doxorubicin, vincristine and prednisolone (CHOP). A post-chemotherapy CT scan revealed no residual disease (Fig. 1. d). He then underwent a course of consolidated intensity-modulated radiotherapy to the surgical bed to a dose of 50Gy in 25 fractions. Post-radiotherapy, the patient received two more cycles of CHOP. The patient tolerated treatment well without significant side effects. At the 26 months follow up point, the patient remains clinically and symptomatically disease-free.
Discussion
Primary adrenal lymphoma or lymphoma involving mainly the adrenal gland without the regional lymph node involvement is a very rare extra-nodal lymphoma. Patients with adrenal lymphoma may present with systemic symptoms including fever, fatigueand weight loss [9–11]. Primary adrenal lymphoma can be discovered incidentally on abdominal imaging. Differential diagnoses include cortical adenoma, pheochromocytoma, metastatic-disease, and adrenocorticotropic hormone (ACTH) dependent Cushing’s syndrome [12, 13]. The definitive diagnosis of primary adrenal lymphoma is made by an image-guided percutaneous biopsy, surgical exploration, or a postmortem examination [1, 2, 14]. Histopathologically, the most common type of primary adrenal lymphoma is B-cell type, mainly diffuse large B-cell lymphoma. Although involvement of the adrenal glands can be seen in disseminated disease, primary NK/T cell lymphoma in the adrenal gland as an initial presentation is extremely rare. The NK/T cell lymphomas are aggressive lymphoproliferative disorders derived from either activated NK cells or cytotoxic T cells. The 2008 World Organization classification system categorized NK–cell malignancies into three types: extra-nodal NK/T-cell lymphoma (nasal and non-nasal), aggressive NK-cell leukemia and chronic lymphoproliferative disorder of NK cells [15]. NK/T-cell lymphomas demonstrate vascular destruction with prominent necrosis. Immunophenotypically, they express CD2 and CD56. NK/T-cell lymphomas are strongly associated with Epstein-Barr virus (EBV), suggesting a pathogenic role of this virus [16, 17]. In this case, the patient’s tumor was positive for CD2, CD56 and EBER.
NK/T cell lymphoma is an extra-nodal lymphoma with predilection for the nasal cavity, nasopharynx, skin, and the other extra-nodal sites. The primary adrenal NK/Tcell lymphoma is exceedingly uncommon. In the review of the literature, we found only seven reported cases of NK/T cell lymphoma presenting as adrenal masses. Among these cases, the adrenal mass was the only involved site in one case [9]. The rest involved multiple organs, including central nervous system (CNS), paranasal sinus, kidney, bone marrow, etc. [1, 3–7]. Tissue diagnosis was made from the adrenal mass in four cases through adrenalectomy, CT-guided biopsy and autopsy specimen [1–3, 16], from cervical lymph nodes or nasal mass in two cases. There was no pathologic diagnosis in one case (the diagnosis was made per imaging study). The ages of the patients ranged from 17 to 76. Among these cases, the highly aggressive nature of the lymphoma and its rapid clinical progression were reported and the long-term prognosis has not been observed (Table. 1).
Because of the rarity of this disease, the standard therapies have not been defined. The treatments have included various combinations of surgery, chemotherapy, and radiation. In the reported cases, four patients received chemotherapy, one patient received surgery and chemotherapy; one patient received high dose intravenous dexamethasone, and one patient received immunosuppression therapy. CHOP was the most common used chemotherapy regimen in these reported cases. The longest reported survival was 90 days in a patient who received surgery and chemotherapy. Our patient underwent surgery, postoperative chemotherapy with 4 cycles of CHOP followed by radiotherapy. Two more cycles of CHOP were administered afterwards. It should be noted that the surgery was performed prior to the patient’s visit at our hospital. If a biopsy sufficed, then surgery would not be necessary, given that these tumors are generally sensitive to chemotherapy and radiation therapy. Because of the residual tumor at the left side of the retroperitoneum, postoperative radiation therapy was recommended. In terms of chemotherapy regimen, SMILE (dexamethasone, methotrexate, ifosfamide, L-asparaginase and etoposide) is a standard chemotherapy regimen for NK/T lymphoma patients in the western countries. However, there was a shortage of L-asparaginase in our province in the end of 2013, when this patient was first treated. Therefore, this patient was treated with CHOP according to the previous case reports [2, 5, 7]. As a side note, SMILE has been used for NK/T lymphoma patients in our province since mid-2014.
Our patient remains disease-free with a follow up of over two years. We list three possible reasons for the longer disease-free survival for our patient: (1) the NK/T cell lymphoma only involved one adrenal gland; (2) the NK/T cell lymphoma did not involve other organs, except for the adjacent kidney; and (3) the utilization of trimodality therapy.
Conclusion
In conclusion, primary adrenal NK/T cell lymphoma is an exceedingly uncommon neoplasm and the outcomes for it are generally poor. We present the first case of a primary adrenal NK/T cell lymphoma in China with over 2-year disease-free survival treated with trimodality therapy. NK/T cell lymphoma should be considered in the differential diagnosis of a rapidly enlarging adrenal mass. As reports of such cases increase and our understanding of the disease improves, we anticipate more effective treatment for these patients.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- CHOP:
-
Cyclophosphamide, doxorubicin, vincristine and prednisolone
- CISH:
-
Chromogenic in situ hybridization
- CNS:
-
Central nervous system
- CT:
-
Computed tomography
- EBV:
-
Epstein-Barr virus
- MRI:
-
Magnetic resonance imaging
- NK/T cell:
-
Natural killer/T cell
- SMILE:
-
dexamethasone, methotrexate, ifosfamide, L-asparaginase and etoposide
References
Dunning KK, Wudhikarn K, Safo AO, Holman CJ, McKenna RW, Pambuccian SE. Adrenal extranodal NK/T-cell lymphoma diagnosed by fine-needle aspiration and cerebrospinal fluid cytology and immunophenotyping: a case report. Diagn Cytopathol. 2009;37(9):686–95.
Thompson MA, Habra MA, Routbort MJ, Holsinger FC, Perrier ND, Waguespack SG, et al. Primary adrenal natural killer/T-cell nasal type lymphoma: first case report in adults. Am J Hematol. 2007;82(4):299–303.
Mizoguchi Y, Nakamura K, Miyagawa S, Nishimura S, Arihiro K, Kobayashi M. A case of adolescent primary adrenal natural killer cell lymphoma. Int J Hematol. 2005;81(4):330–4.
Toba A, Tamura Y, Osajima Y, Kinbara Y, Sato M, Yamaga R. A case of nasal NK/T cell lymphoma presenting with bilateral giant adrenal tumors [in Japanese]. Nippon Ronen IgakkaiZasshi. 2008;45(6):660–5.
Nagireddy S, Avery RA, Kakivayi S, Matin K. Natural killer T-cell lymphoma presenting as adrenal mass and partial adrenal insufficiency followed by CNS involvement. J Clin Oncol. 2011;29(15):e431–3.
Kang SM, Kim WJ, Lee KA, Baek HS, Park TS, Jin HY. Mainly adrenal gland involving NK/T-cell nasal type lymphoma diagnosed with delay due to mimicking adrenal hemorrhage. J Korean Med Sci. 2011;26(10):1386–90.
Jin Juying Y. yerong. NK/T cell lymphoma involving bilateral adrenal glands: one case report [in Chinese]. Chin J. Endocrinol Metab. 2005;21(1):82.
AlShemmari SH, Ameen RM, Sajnani KP. Extranodal lymphoma: a comparative study. Hematology. 2008;13(3):163–9.
Mantzios G, Tsirigotis P, Veliou F, Boutsikakis I, Petraki L, Kolovos J, et al. Primary adrenal lymphoma presenting as Addison’s disease: case report and review of the literature. Ann Hematol. 2004;83(7):460–3.
Levy NT, Young Jr WF, Habermann TM, Strickler JG, Carney JA, Stanson AW. Adrenal insufficiency as a manifestation of disseminated non-Hodgkin’s lymphoma. Mayo Clin Proc. 1997;72(9):818–22.
Grigg AP, Connors JM. Primary adrenal lymphoma. Clin Lymphoma. 2003;4(3):154–60.
Arora S, Vargo S, Lupetin AR. Computed tomography appearance of spontaneous adrenal hemorrhage in a pheochromocytoma. Clin Imaging. 2009;33(4):314–7.
Nawar R, Aron D. Adrenal incidentalomas - a continuing management dilemma. EndocrRelat Cancer. 2005;12(3):585–98.
Tomoyose T, Nagasaki A, Uchihara JN, Kinjo S, Sugaya K, Onaga T, et al. Primary adrenal adult T-cell leukemia/lymphoma: a case report and review of the literature. Am J Hematol. 2007;82(8):748–52.
Chan JKC, Quintanilla-Martinez L, Ferry JA, Peh SC. Extranodal NK/T-cell lymphoma, nasal type. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., editors. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. p. 285–8.
Kanavaros P, Lescs MC, Brière J, Divine M, Galateau F, Joab I, et al. Nasal T-cell lymphoma: A clinicopathologic entity associated with peculiar phenotype and with Epstein-Barr virus. Blood. 1993;81(10):2688–95.
Ko YH, Ree HJ, Kim WS, Choi WH, Moon WS, Kim SW. Clinicopathologic and genotypic study of extranodal nasal-type natural killer/T-cell lymphoma and natural killer precursor lymphoma among Koreans. Cancer. 2000;89(10):2106–16.
Acknowledgements
None.
Funding
Applied Basic Research Program in Wuhan (Grant No. 2015060101010054); Natural Science Foundation of Hubei (Grant No. 2016CFC737).
Availability of data and materials
All relevant data are within the manuscript.
Authors’ contributions
LH and WMX drafted the manuscript and performed the literature search; MWW carried out pathological examination and contributed to the histopathological details of the manuscript; PW acquired the clinical data and was responsible for interpretation of the data; GH and CL conceived the idea for this case report, referred to the literature review aspect of this case report and revised the article for important intellectual content. All authors were responsible for critical revision of the manuscript. All authors have read and approved the final manuscript.
Authors’ information
Liu Hu, Radiation Oncologist
Weimin Xu, Surgeon
Mingwei Wang, Pathologist
Pan Wang, Physician
Guang Han, Radiation Oncologist
Chi Lin, Radiation Oncologist
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the editor of this journal.
Ethics approval and consent to participate
Not applicable.
CARE guidelines/methodology
The authors adhered to CARE guidelines/methodology.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, L., Xu, W., Wang, M. et al. A case report of primary unilateral adrenal NK/T cell lymphoma: good clinical outcome with trimodality treatment. BMC Cancer 17, 15 (2017). https://doi.org/10.1186/s12885-016-3019-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-016-3019-1