
Abstract
Background
Chronic Myeloid Leukemia was always referred as a unique cancer due to the apparent independence from tumor suppressors’ deletions/mutations in the early stages of the disease. However, it is now well documented that even genetically wild-type tumor suppressors can be involved in tumorigenesis, when functionally inactivated. In particular, tumor suppressors’ functions can be impaired by subtle variations of protein levels, changes in cellular compartmentalization and post-transcriptional/post-translational modifications, such as phosphorylation, acetylation, ubiquitination and sumoylation. Notably, tumor suppressors inactivation offers challenging therapeutic opportunities. The reactivation of an inactive and genetically wild-type tumor suppressor could indeed promote selective apoptosis of cancer cells without affecting normal cells.
Main body
Chronic Myeloid Leukemia (CML) could be considered as the paradigm for non-genomic loss of function of tumor suppressors due to the ability of BCR-ABL to directly promote functionally inactivation of several tumor suppressors.
Short conclusion
In this review we will describe new insights on the role of FoxO, PP2A, p27, BLK, PTEN and other tumor suppressors in CML pathogenesis. Finally, we will describe strategies to promote tumor suppressors reactivation in CML.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Chronic Myeloid Leukemia (CML) was generally referred as an unique cancer, due to the apparent independence from tumor suppressors’ deletions/mutations in the early stages of the disease [1]. In agreement with this concept, infection of murine stem cells with BCR-ABL-expressing vectors was also associated with rapid development of CML without the need of additional genetic lesions [2]. Over the last few years, the involvement of tumor suppressors (TS) in cancer pathogenesis has been completely revised [3–5]. In particular, while in the original Knudson’s model TS are involved in tumorigenesis upon inactivation of both alleles (generally one through point mutation and one through deletion), it is now clear that even genetically wild-type TS can modulate tumorigenesis when down-regulated, aberrantly compartmentalized and/or affected by phosphorylation/acetylation/ubiquitination and others post transcriptional modifications.
In line with these observations, CML could represent the paradigm of how cancer can arise upon functional inactivation of tumor suppressors. In this review, we will describe how BCR-ABL directly promotes TS inactivation with important therapeutic implications. Finally, we will also describe those TS that are inactive in CML but without a clear direct regulation by BCR-ABL.
Tumor suppressors directly inactivated by BCR-ABL
FoxO
The Forkhead box subgroup O (FoxO) family of transcription factors (TFs) is a subclass of Forkhead transcription factors characterized by a winged helix DNA binding domain known as a Forkhead box [6, 7]. This family comprises four members (FoxO1, FoxO3, FoxO4 and FoxO6). In the presence of several Growth Factors (GFs) or activated tyrosine kinases, the PI3K-AKT signal transduction pathway promotes FoxO phosphorylation, favoring nuclear exclusion and suppression of transcriptional activity. Conversely, in the absence of GFs, un-phosphorylated FoxOs translocate into the nucleus where they modulate the expression of several genes. Furthermore, FoxOs are regulated by several protein modifications, such as acetylation, ubiquitination and arginine/lysine methylation. FoxOs have been described as essential components of BCR-ABL signal transduction [8–10]. In particular, BCR-ABL is a strong activator of the PI3K-AKT pathway and therefore promotes the inactivation of FoxO3a, FoxO1 and FoxO4 though phosphorylation and shuttling into the cytoplasm. On the contrary, Tyrosine Kinase Inhibitor (TKI) treatment promotes the reactivation of FoxOs which in turn are able to mediate cell cycle arrest. Reactivation of FoxOs is associated with the down-regulation of CCND1/Cyclin D1 protein expression and affects the expression of stem cell genes such as ATM, p57/CDKN1C, and BCL6 [10]. Similarly, another report highlighted BCL6 as an essential FoxO downstream mediator of cell renewal [11]. As a consequence, FoxOs reactivation impacts on the maintenance of the leukemia stem cells without affecting the normal hemopoietic stem cell compartment. Other authors have also shown that TGF-beta is involved in the regulation of FoxOs with consequent regulation of the LSC compartment [9, 12]. Notably, the BCR ABL/PI3K/AKT/FoxO pathway is less dependent on BCR-ABL activity in the stem cell compartment [10]. This finding could explain the reason why stem cells remain quiescence even in the presence of BCR-ABL and are resistant to TKI treatment. The mechanism of FoxOs nuclear retention in stem cells is still not explained in detail, although it was associated with AKT-mediated phosphorylation. Since FoxOs localization is also regulated by mono-ubiquitination [13] and that BCR-ABL activates the FoxOs-deubiquitinase HAUSP [14], it could be speculated that FoxOs nuclear localization could be affected by BCR-ABL/PML/HAUSP network in a similar manner as for PTEN [14]. However, experimental studies are mandatory to demonstrate this network with important therapeutic implications, due to the availability of HAUSP inhibitors (Fig. 1).

Tumor suppressors network associated with the BCR-ABL/PI3K/AKT pathway. Schematic representation of the BCR-ABL/PI3K/AKT pathway and the role of PTEN and FOXO tumor suppressors. This carton highlights how BCR-ABL inactivates PTEN through CKII-mediated phosphorylation and HAUSP-mediated changes of cellular compartmentalization. Furthermore, BCR-ABL promotes FOXO inactivation through the regulation of its cellular localization. We also speculate on the putative regulation of FOXO localization through HAUSP in CML, although BCR-ABL/HAUSP/FOXO connection has to be demonstrated
PP2A
In the last years, it was demonstrated that BCR-ABL, irrespectively to its tyrosine kinase activity, is able to promote the recruitment and the activation of the Janus kinase 2 (JAK2) [15]. JAK2 is in turn able to enhance β-catenin activity which is responsible of SET-mediated inactivation of protein phosphatase 2A (PP2A). PP2A is a ubiquitous serine/threonine phosphatase that targets Raf, MEK, AKT and other essential mediators of oncogenic signals [16]. Besides having linked β-catenin signaling to the inactivation of a tumor suppressor, the relevance of these observations relies on the fact that PP2A activity can be restored by PP2A activating drugs [17]. In particular, the orally available FTY720 promotes the activation of PP2A favoring CML cells and CML stem cells apoptosis [18, 19]. Most importantly, this drug was shown to induce apoptosis in the tyrosine kinase resistant stem cell pool [19] (Fig. 2).

Tumor suppressors network associated with the BCR-ABL/MAPK pathway. Representation of the tumor suppressors involved in the RAS/MEK/ERK pathway and the JAK2/β-catenin pathway. DOK family proteins are involved in the negative regulation of RAS activation. Representation of the IRF pathway. Finally, morgana/chp-1 pathway is indicated
p27
p27 is an inhibitor of cyclin-dependent kinases (Cdk2) involved in the control of cell-cycle [20]. As most of the regulator of cell-cycle, p27 is tightly regulated at different levels. P27 has been referred as a tumor suppressor, although a paradoxical dual role (oncogenic/tumor suppressor role) has been postulated. Notably, changes in p27 cellular compartmentalization appears to play an essential role in tumorigenesis: nuclear exclusion was indeed associated with adverse prognosis in several cancers [21]. BCR-ABL was shown to regulate p27 at different levels. In particular, BCR-ABL affects p27 expression and promotes degradation of nuclear p27 [22–25]. Moreover, BCR-ABL promotes FoxO3a inhibition through PI3K-AKT with consequent impairment of p27 transcription. Furthermore, PI3K regulates the activity of SKP2 which mediates p27 degradation. BCR-ABL is also able to promote p27 phosphorylation on tyrosine 88 which is involved in the control of cyclinE/Cdk2 activity. More recently, BCR-ABL was shown to promote oncogenic gain of functions of cytoplasmic p27 [26]. The overall role of p27 in CML pathogenesis is that nuclear p27 acts as a tumor suppressor promoting cell cycle regulation; on the contrary, cytoplasmic p27 is acting as an oncogene. The relevance of p27 network relies on the fact that forcing p27 into the nucleus can dictate cancer selective growth arrest and apoptosis [26] (Fig. 3).

BCR-ABL/p53 connection, p27 network and miRNAs in CML. BCR-ABL promotes either sequestration of p53 in the cytoplasm through the interaction with IkB-alpha and the p53 degradation through MDM2. Furthermore, the interaction BCR-ABL with p27 is reported
PTEN
The tumor suppressor PTEN is involved in either the regulation of the PI3K-AKT pathway and phosphatase independent functions [27]. Several recent reports have demonstrated that PTEN plays an essential role in the pathogenesis of CML [28], as reviewed elsewhere [29]. In particular, PTEN was reported to be i) under-expressed through a Ras-MEK pathway [30, 31], ii) inactivated through tail phosphorylation [32] and iii) delocalized into the cytoplasm [14] (Fig. 1).
DOK genes
DOK1 and DOK2 are rasGAP-associated docking proteins, that are preferentially expressed in the hematopoietic cells, and behave as tumor suppressors in both myeloproliferative disorders and lung cancer [33]. DOK proteins contain a NH2-terminal Pleckstrin homology domain (PH), a Phosphotyrosine-binding domain (PTB) and a COOH-terminal SH2 target motif. DOK proteins bind to p120 rasGAP and therefore counteract the activation of the RAS-RAF-MEK pathway. DOK1, also known as p62dok, and DOK2 were originally cloned as a BCR-ABL substrate in CML [34–37]. Expression of both Dok1 and Dok2 opposes BCR-ABL mediated leukemogenesis [38, 39]. Although DOK1 and DOK2 have not been extensively studied in human CML samples, it was demonstrated that DOK phosphorylation by BCR-ABL is associated with the inactivation of its activity as a Ras-GAP [40]. Furthermore, BCR-ABL was also shown to promote DOK1 ubiquitination and degradation [41]. All together, these data indicate that DOK proteins act as tumor suppressors through the inhibition of the RAS-MEK-ERK pathway, but in CML their function is directly inhibited by BCR-ABL (Fig. 2).
p53
TP53 is a tumor suppressor exerting a pivotal role for the maintenance of genomic integrity in response to several cellular stresses [42]. According to the damage, p53 induces the transcription of several genes that block cell cycle, or that promote apoptosis, like p21/WAF1 and Bax [43]. The p53 protein is generally expressed at low levels in normal cells and has a short half-life [44]. P53 function is counteracted by MDM2 oncoprotein, that by binding the p53 transactivation domain, inhibits its transcriptional activity, and promotes p53 nuclear export. Moreover, MDM2 acts like a E3 ubiquitin ligase, mediating p53 degradation in a proteasome-dependent manner. Furthermore MDM2 gene is a direct transcriptional target of p53, thereby p53 and MDM2 form a negative feedback loop where p53 regulates the expression of MDM2, that in turn blocks p53 functions and promotes its degradation. The tumor suppressor TP53 plays an essential role in the pathogenesis of several cancers. Within myeloid malignancies, TP53 was also implicated in the progression of CML into the blast phase [45]. In particular, almost 20 % of CML blast phases express TP53 mutations, but no mutations/deletions were reported in the chronic phase of CML. Although un-mutated and not deleted, p53 is functionally inactivated in the chronic phase of CML patients [46]. A mechanism described by Calabretta’s group shows that BCR-ABL upregulates the expression of MDM2 by increasing its translation that is dependent on high levels of the La antigen, an RNA binding protein. The BCR-ABL/MDM2 regulation could indeed affect p53 function. P53 activity could also be regulated by the phospho-status of its negative regulator MDM2. AKT-mediated phosphorylation of MDM2 promotes its nuclear localization that favors the inhibition of p53 [47]. Recently, we have shown that BCR-ABL is able to stabilize an IkB-alpha/p53 complex which is responsible for the sequestration of p53 into the cytoplasm of CML cells [48]. In particular, the NF-kB inhibitor IkB-alpha is able to interact with either NF-kB p65 subunit or the p53 protein. This complex prevents p53 to interact with DNA response elements and to promote apoptosis. Notably, BCR-ABL is able to interact and stabilize IkB-alpha in the cytoplasm therefore promoting p53 sequestration into the cytosol. As a consequence, IkB-alpha prevents p53 mediated apoptosis (Fig. 3).
IRF-8 and IRF-4
The interferon regulatory factor-8 (IRF-8) is an essential myeloid transcription factor involved in the regulation of the myeloid lineage commitment [49]. Notably, IRF-8 deletion in the mouse is associated with the development of CML like MPD [50]. Interestingly, IRF-8 is under-expressed in CML [51]. BCR-ABL activates STAT5 which in turn represses IRF-8. The BCR-ABL/STAT5/IRF-8 network is another example of the BCR-ABL ability to promote tumor suppressors inactivation. Similarly, BCR-ABL regulates the function of another interferon regulatory factor (IRF-4), suggesting that these transcription factors are downstream effectors of the chimeric translocation [52] (Fig. 2).
BCR-ABL/oncogenic miRNA mediated tumor suppressors down-regulation
The involvement of miRNAs in CML pathogenesis is highly complex and include both oncogenic miRNA and tumor suppressive miRNAs [53]. In line with the aim of this review, it should be noted that BCR-ABL is able to positively regulate several oncogenic miRNAs which in turn affect the expression of tumor suppressors [54], with consequent inactivation.
In line with these considerations, for instance, BCR-ABL is able to regulate the expression of oncogenic miR-130a and miR-130b which in turn affect the expression of the tumor suppressor CCN3 [55].
Tumor suppressors, involved in CML pathogenesis, not directly regulated by BCR-ABL
In this section, we will report on tumor suppressors that have been described as inactive in CML, although in a genetically wild-type status. In particular, we focus on those tumor suppressors that are not directly regulated by BCR-ABL but that cooperate with BCR-ABL in the development of CML.
Morgana/chp-1
Morgana/chp-1 is a chaperon protein involved in the regulation of centrosome duplication and genomic stability [56]. Morgana forms a complex with ROCKI and ROCKII promoting the inhibition of their kinase activity and therefore suppressing centrosome over-duplication. We have recently demonstrated that morgana haploinsufficiency is associated with the development of a transplantable myeloproliferative disorder [57]. Furthermore, we have observed that a portion of CML exhibits morgana under-expression, which is associated with increased centrosome amplification and aneuploid metaphases. Notably, patients expressing low levels of morgana are associated with a worse response to TKIs. Due to the ability of morgana to regulate ROCK activity, the sensitivity to TKI of cell obtained from morgana underexpressing patients can be rescued by treating cells with ROCK inhibitors. These data suggest that morgana/chp-1 can cooperate with BCR-ABL in the pathogenesis of CML and in the development of TKI resistant CML. However, the mechanisms of morgana downregulation in CML still have to be clarified (Fig. 2).
BLK
By using microarray analyses of leukemic stem cells, it was showed that the Blk gene is markedly down-regulated in the CML stem cell pool. BLK expression was dependent on BCR-ABL protein but independent of its kinase activity [58]. Notably, Blk was shown to be involved in the regulation of Leukemic stem cells maintenance. Blk is a member of the Src tyrosine kinase. Although Src proteins behave as oncogenes, Blk was shown to act as a tumor suppressor through the regulation of CML cells proliferation, in a pathway involving c-myc and p27 (Fig. 3).
Tumor suppressive miRNAs
Various miRNAs with known tumor suppressive roles have been found de-regulated in CML. In particular, miR-29a and miR-29b were shown to be down-modulated in CML and anti-correlated with the expression levels of target genes, Bcl-2 and Mcl-1 [59]. Interestingly, miR-424 and miR-320a that directly target the 3′UTR of the ABL gene are under-expressed in CML and miR-320a is also downregulated in CML cancer stem cells [60, 61]. Up-regulation of these miRNAs inhibits cell proliferation, induces apoptosis and, in the specific case of miR-424, also increases the sensibility to the Imatinib treatment. Others miRNAs have also been involved in the pathogenesis of CML [61, 62]. However, it should be noted that further analyses should be performed to address the mechanisms of miRNA deregulation in CML and the real in vivo contribution in CML pathogenesis (Fig. 3).
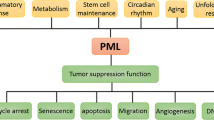
PML
The tumor suppressor PML plays an essential role in the regulation of CML stem cell [63], and various reviews have been published on this topic [64, 65]. Furthermore, PML plays an essential role in the regulation of the tumor suppressive function of PTEN, through HAUSP [66]. The tumor suppressive functions of PML in CML are associated with the differential PML expression during the leukemic differentiation. While PML retains high levels of expression in the stem cell compartment, where it mediates stem cell quiescence, PML levels progressively drop during differentiation into progenitor and terminally differentiated cells. As a consequence, loss of PML is associated with both increased proliferation [63] and PTEN nuclear pool exclusion [14]. While the mechanism of PML tumor suppressive functions in CML are highly complex, it should be noted that PML is a targetable tumor suppressor due to the ability of arsenic trioxide to promote its degradation. Even if apparently contradictory in the context of cancer therapy, the degradation of PML promotes cell cycle induction of CML stem cells with consequent their exhaustion. PML targeting strategies offer the chance to achieve the eradication of CML [63].
Strategies to promote tumor suppressor reactivation
The inability to overcome genetic inactivation of tumor suppressors with anticancer therapies is currently challenging. Conversely, targeting mechanisms implicated in non genomic tumor suppressor loss of function could become a new potential strategy to enhance or support target therapy responses. In particular, inhibitors of CKII are able to promote PTEN tumor suppressive functions [32]. Accordingly, HAUSP inhibitors, as well as arsenic trioxide [63] could restore PTEN nuclear localization with pro-apoptotic and antiproliferative effects. Similarly, reactivation of PP2A was show to antagonize oncogenic BCR-ABL properties in vitro [17]. Direct pharmacological activation of PP2A by Forskolin, or indirect targeting of inhibitor components of PPA2 pathway (such as SET inhibitors) reduced proliferation and clonogenic potential, and induced apoptosis in myeloid malignances [19]. The loss of a tumor suppressor gene can also cause the activation of a side pathway. This is what happens in CML patients expressing low levels of Morgana. The increase of ROCK activity consequent to Morgana modulation confers imatinib resistance. Treatment with ROCK inhibitor was shown to rescue the apoptotic response to imatinib [57].
Discussion
The oncogenic BCR-ABL signal is part of a complex network of interactions that mediate proliferation and survival. Parallel to these signaling transduction pathways, BCR-ABL is also able to mediate the inactivation of several tumor suppressors, through either i) regulation of gene expression, or ii) changes in cellular compartmentalization or iii) directly or indirectly, favoring protein modifications, such as phosphorylation/ubiquitination/acetylation. The relevance of these networks relies on the fact that targeting mechanisms that promote tumor suppressors inactivation can restore their function with consequent strong and selective cancer apoptosis. In our opinion, the development of strategies to reactivate tumor suppressors is a really challenging therapeutic option and CML could represent an essential model to verify the efficacy of this novel targeted molecular therapy. In particular, those cases characterized by resistance to TKI could benefit with combined therapy to achieve synthetic lethality [67].
Conclusion
CML chronic phase is not associated with known TS genetic loss of function, suggesting that BCR-ABL is sufficient for the development of this disease. However, as we have reviewed here, BCR-ABL has the ability to functionally inactivate several tumor suppressors allowing to promote tumorigenesis through an highly complex signal transduction network. The functional inactivation of TS is a great opportunity to design combinatorial therapies to achieve synthetic lethality together with BCR-ABL tyrosine kinase inhibitors.
Abbreviations
- CML:
-
chronic myeloid leukemia
- TS:
-
tumor suppressors
- FoxO:
-
Forkhead box subgroup O
- GF:
-
growth factors
- TKI:
-
tyrosine kinase inhibitors
- JAK2:
-
Janus kinase 2
- PTB:
-
phosphotyrosine-binding domain
- IRF-8:
-
interferon regulatory factor-8
References
Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–53.
Van Etten RA. Studying the pathogenesis of BCR-ABL+ leukemia in mice. Oncogene. 2002;21:8643–51.
Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476:163–9.
Correia NC, Gírio A, Antunes I, Martins LR, Barata JT. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur J Cancer Oxf Engl 1990. 2014;50:216–25.
Leslie NR, Foti M. Non-genomic loss of PTEN function in cancer: not in my genes. Trends Pharmacol Sci. 2011;32:131–40.
Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39:159–69.
Lam EW-F, Brosens JJ, Gomes AR, Koo C-Y. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13:482–95.
Jagani Z, Singh A, Khosravi-Far R: FoxO tumor suppressors and BCR-ABL-induced leukemia: a matter of evasion of apoptosis. Biochim Biophys Acta 2008, 1785:63–84
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–80.
Pellicano F, Scott MT, Helgason GV, Hopcroft LEM, Allan EK, Aspinall-O’Dea M, Copland M, Pierce A, Huntly BJP, Whetton AD, Holyoake TL. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells Dayt Ohio. 2014;32:2324–37.
Hurtz C, Hatzi K, Cerchietti L, Braig M, Park E, Kim Y, Herzog S, Ramezani-Rad P, Jumaa H, Müller MC, Hofmann W-K, Hochhaus A, Ye BH, Agarwal A, Druker BJ, Shah NP, Melnick AM, Müschen M. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med. 2011;208:2163–74.
Naka K, Hoshii T, Hirao A. Novel therapeutic approach to eradicate tyrosine kinase inhibitor resistant chronic myeloid leukemia stem cells. Cancer Sci. 2010;101:1577–81.
van der Horst A, de Vries-Smits AMM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BMT. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8:1064–73.
Morotti A, Panuzzo C, Crivellaro S, Pergolizzi B, Familiari U, Berger AH, Saglio G, Pandolfi PP. BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia. 2014;28:1326–33.
Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, Roy DC, Valtieri M, Bruner-Klisovic R, Caligiuri MA, Bloomfield CD, Marcucci G, Perrotti D. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–68.
Haesen D, Sents W, Lemaire K, Hoorne Y, Janssens V. The Basic Biology of PP2A in Hematologic Cells and Malignancies. Front Oncol. 2014;4:347.
Ciccone M, Calin GA, Perrotti D. From the Biology of PP2A to the PADs for Therapy of Hematologic Malignancies. Front Oncol. 2015;5:21.
Kiyota M, Kuroda J, Yamamoto-Sugitani M, Shimura Y, Nakayama R, Nagoshi H, Mizutani S, Chinen Y, Sasaki N, Sakamoto N, Kobayashi T, Matsumoto Y, Horiike S, Taniwaki M. FTY720 induces apoptosis of chronic myelogenous leukemia cells via dual activation of BIM and BID and overcomes various types of resistance to tyrosine kinase inhibitors. Apoptosis Int J Program Cell Death. 2013;18:1437–46.
Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, Ferenchak G, Dorrance AM, Paisie CA, Eiring AM, Ma Y, Mao HC, Zhang B, Wunderlich M, May PC, Sun C, Saddoughi SA, Bielawski J, Blum W, Klisovic RB, Solt JA, Byrd JC, Volinia S, Cortes J, Huettner CS, Koschmieder S, Holyoake TL, Devine S, Caligiuri MA, Croce CM, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123:4144–57.
Roy A, Banerjee S. p27 and leukemia: cell cycle and beyond. J Cell Physiol. 2015;230:504–9.
Rosen DG, Yang G, Cai KQ, Bast RC, Gershenson DM, Silva EG, Liu J. Subcellular localization of p27kip1 expression predicts poor prognosis in human ovarian cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11(2 Pt 1):632–7.
Jonuleit T, van der Kuip H, Miething C, Michels H, Hallek M, Duyster J, Aulitzky WE. Bcr-Abl kinase down-regulates cyclin-dependent kinase inhibitor p27 in human and murine cell lines. Blood. 2000;96:1933–9.
Gesbert F, Sellers WR, Signoretti S, Loda M, Griffin JD. BCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-Kinase/AKT pathway. J Biol Chem. 2000;275:39223–30.
Andreu EJ, Lledó E, Poch E, Ivorra C, Albero MP, Martínez-Climent JA, Montiel-Duarte C, Rifón J, Pérez-Calvo J, Arbona C, Prósper F, Pérez-Roger I. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27Kip1 degradation and proliferation of chronic myelogenous leukemia cells. Cancer Res. 2005;65:3264–72.
Agarwal A, Bumm TGP, Corbin AS, O’Hare T, Loriaux M, VanDyke J, Willis SG, Deininger J, Nakayama KI, Druker BJ, Deininger MW. Absence of SKP2 expression attenuates BCR-ABL-induced myeloproliferative disease. Blood. 2008;112:1960–70.
Agarwal A, Mackenzie RJ, Besson A, Jeng S, Carey A, LaTocha DH, Fleischman AG, Duquesnes N, Eide CA, Vasudevan KB, Loriaux MM, Firpo E, Cortes JE, McWeeney S, O’Hare T, Roberts JM, Druker BJ, Deininger MW. BCR-ABL1 promotes leukemia by converting p27 into a cytoplasmic oncoprotein. Blood. 2014;124:3260–73.
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96.
Peng C, Chen Y, Yang Z, Zhang H, Osterby L, Rosmarin AG, Li S. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood. 2010;115:626–35.
Morotti A: The role of the tumor suppressor PTEN in Chronic Myeloid Leukemia pathogenesis. Sci Proc. 2015;2:e638. doi:10.14800/sp.638.
Panuzzo C, Crivellaro S, Carrà G, Guerrasio A, Saglio G, Morotti A. BCR-ABL promotes PTEN downregulation in chronic myeloid leukemia. PloS One. 2014;9:e110682.
Huang F-F, Zhang L, Wu D-S, Yuan X-Y, Yu Y-H, Zhao X-L, Chen F-P, Zeng H. PTEN regulates BCRP/ABCG2 and the side population through the PI3K/Akt pathway in chronic myeloid leukemia. PloS One. 2014;9:e88298.
Morotti A, Panuzzo C, Crivellaro S, Carrà G, Fava C, Guerrasio A, Pandolfi PP, Saglio G. BCR-ABL inactivates cytosolic PTEN through Casein Kinase II mediated tail phosphorylation. Cell Cycle Georget Tex. 2015;14:973–9.
Berger AH, Niki M, Morotti A, Taylor BS, Socci ND, Viale A, Brennan C, Szoke J, Motoi N, Rothman PB, Teruya-Feldstein J, Gerald WL, Ladanyi M, Pandolfi PP. Identification of DOK genes as lung tumor suppressors. Nat Genet. 2010;42:216–23.
Master Z, Tran J, Bishnoi A, Chen SH, Ebos JML, Van Slyke P, Kerbel RS, Dumont DJ. Dok-R binds c-Abl and regulates Abl kinase activity and mediates cytoskeletal reorganization. J Biol Chem. 2003;278:30170–9.
Cristofano AD, Carpino N, Dunant N, Friedland G, Kobayashi R, Strife A, Wisniewski D, Clarkson B, Pandolfi PP, Resh MD. Molecular Cloning and Characterization of p56 dok-2 Defines a New Family of RasGAP-binding Proteins. J Biol Chem. 1998;273:4827–30.
Di Cristofano A, Niki M, Zhao M, Karnell FG, Clarkson B, Pear WS, Van Aelst L, Pandolfi PP. p62(dok), a negative regulator of Ras and mitogen-activated protein kinase (MAPK) activity, opposes leukemogenesis by p210(bcr-abl). J Exp Med. 2001;194:275–84.
Carpino N, Wisniewski D, Strife A, Marshak D, Kobayashi R, Stillman B, Clarkson B. p62(dok): a constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 1997;88:197–204.
Yasuda T, Shirakata M, Iwama A, Ishii A, Ebihara Y, Osawa M, Honda K, Shinohara H, Sudo K, Tsuji K, Nakauchi H, Iwakura Y, Hirai H, Oda H, Yamamoto T, Yamanashi Y. Role of Dok-1 and Dok-2 in myeloid homeostasis and suppression of leukemia. J Exp Med. 2004;200:1681–7.
Niki M, Di Cristofano A, Zhao M, Honda H, Hirai H, Van Aelst L, Cordon-Cardo C, Pandolfi PP. Role of Dok-1 and Dok-2 in leukemia suppression. J Exp Med. 2004;200:1689–95.
Kashige N, Carpino N, Kobayashi R. Tyrosine phosphorylation of p62dok by p210bcr-abl inhibits RasGAP activity. Proc Natl Acad Sci U S A. 2000;97:2093–8.
Janas JA, Van Aelst L. Oncogenic tyrosine kinases target Dok-1 for ubiquitin-mediated proteasomal degradation to promote cell transformation. Mol Cell Biol. 2011;31:2552–65.
Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–17.
Khoo KH, Hoe KK, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–36.
Tao W, Levine AJ. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc Natl Acad Sci U S A. 1999;96:3077–80.
Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–22.
Trotta R, Vignudelli T, Candini O, Intine RV, Pecorari L, Guerzoni C, Santilli G, Byrom MW, Goldoni S, Ford LP, Caligiuri MA, Maraia RJ, Perrotti D, Calabretta B. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell. 2003;3:145–60.
Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–603.
Crivellaro S, Panuzzo C, Carrà G, Volpengo A, Crasto F, Gottardi E, Familiari U, Papotti M, Torti D, Piazza R, Redaelli S, Taulli R, Guerrasio A, Saglio G, Morotti A. Non genomic loss of function of tumor suppressors in CML: BCR-ABL promotes IκBα mediated p53 nuclear exclusion. Oncotarget. 2015;6:25217–25.
Tamura T, Kurotaki D, Koizumi S. Regulation of myelopoiesis by the transcription factor IRF8. Int J Hematol. 2015;101:342–51.
Watanabe T, Hotta C, Koizumi S, Miyashita K, Nakabayashi J, Kurotaki D, Sato GR, Yamamoto M, Nakazawa M, Fujita H, Sakai R, Fujisawa S, Nishiyama A, Ikezawa Z, Aihara M, Ishigatsubo Y, Tamura T. The transcription factor IRF8 counteracts BCR-ABL to rescue dendritic cell development in chronic myelogenous leukemia. Cancer Res. 2013;73:6642–53.
Waight JD, Banik D, Griffiths EA, Nemeth MJ, Abrams SI. Regulation of the interferon regulatory factor-8 (IRF-8) tumor suppressor gene by the signal transducer and activator of transcription 5 (STAT5) transcription factor in chronic myeloid leukemia. J Biol Chem. 2014;289:15642–52.
Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M, Takami M, Ichino M, Nakazawa M, Matsuyama T, Kamijo R, Kitagawa S, Ozato K, Tamura T. Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PloS One. 2011;6:e25812.
Di Stefano C, Mirone G, Perna S, Marfe G. The roles of microRNAs in the pathogenesis and drug resistance of chronic myelogenous leukemia (Review). Oncol Rep. 2016;35:614–24.
Faber J, Gregory RI, Armstrong SA. Linking miRNA regulation to BCR-ABL expression: the next dimension. Cancer Cell. 2008;13:467–9.
Suresh S, McCallum L, Lu W, Lazar N, Perbal B, Irvine AE. MicroRNAs 130a/b are regulated by BCR-ABL and downregulate expression of CCN3 in CML. J Cell Commun Signal. 2011;5:183–91.
Ferretti R, Palumbo V, Di Savino A, Velasco S, Sbroggiò M, Sportoletti P, Micale L, Turco E, Silengo L, Palumbo G, Hirsch E, Teruya-Feldstein J, Bonaccorsi S, Pandolfi PP, Gatti M, Tarone G, Brancaccio M. Morgana/chp-1, a ROCK inhibitor involved in centrosome duplication and tumorigenesis. Dev Cell. 2010;18:486–95.
Di Savino A, Panuzzo C, Rocca S, Familiari U, Piazza R, Crivellaro S, Carrà G, Ferretti R, Fusella F, Giugliano E, Camporeale A, Franco I, Miniscalco B, Cutrin JC, Turco E, Silengo L, Hirsch E, Rege-Cambrin G, Gambacorti-Passerini C, Pandolfi PP, Papotti M, Saglio G, Tarone G, Morotti A, Brancaccio M. Morgana acts as an oncosuppressor in chronic myeloid leukemia. Blood. 2015;125:2245–53.
Zhang H, Peng C, Hu Y, Li H, Sheng Z, Chen Y, Sullivan C, Cerny J, Hutchinson L, Higgins A, Miron P, Zhang X, Brehm MA, Li D, Green MR, Li S. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat Genet. 2012;44:861–71.
Li Y, Wang H, Tao K, Xiao Q, Huang Z, Zhong L, Cao W, Wen J, Feng W. miR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Exp Cell Res. 2013;319:1094–101.
Hershkovitz-Rokah O, Modai S, Pasmanik-Chor M, Toren A, Shomron N, Raanani P, Shpilberg O, Granot G. Restoration of miR-424 suppresses BCR-ABL activity and sensitizes CML cells to imatinib treatment. Cancer Lett. 2015;360:245–56.
Xishan Z, Ziying L, Jing D, Gang L. MicroRNA-320a acts as a tumor suppressor by targeting BCR/ABL oncogene in chronic myeloid leukemia. Sci Rep. 2015;5:12460.
Fallah P, Amirizadeh N, Poopak B, Toogeh G, Arefian E, Kohram F, Hosseini Rad SMA, Kohram M, Teimori Naghadeh H, Soleimani M. Expression pattern of key microRNAs in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Int J Lab Hematol. 2015;37:560–8.
Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, Pandolfi PP. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–8.
Ito K, Bernardi R, Pandolfi PP. A novel signaling network as a critical rheostat for the biology and maintenance of the normal stem cell and the cancer-initiating cell. Curr Opin Genet Dev. 2009;19:51–9.
Nakahara F, Weiss CN, Ito K. The role of PML in hematopoietic and leukemic stem cell maintenance. Int J Hematol. 2014;100:18–26.
Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455:813–7.
Morotti A, Panuzzo C, Fava C, Saglio G. Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid leukemia. Expert Opin Biol Ther. 2014;14:287–99.
Acknowledgment
This work was supported by the Giovani Ricercatori – Ricerca Finalizzata 2010, code GR-2010-2312984, to A.M.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
Authors declare no conflict of interest regarding this review.
Authors’ contribution
The manuscript was written by AM, GC. and SC. Figures were prepared by GC.; CP, RT, AG and GS reviewed the manuscript and contributed in the planning of the review. All authors have read and approved the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Crivellaro, S., Carrà, G., Panuzzo, C. et al. The non-genomic loss of function of tumor suppressors: an essential role in the pathogenesis of chronic myeloid leukemia chronic phase. BMC Cancer 16, 314 (2016). https://doi.org/10.1186/s12885-016-2346-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-016-2346-6