
Abstract
Background
Fraser syndrome or “cryptophthalmos syndrome” is a rare autosomal recessive disease. It is characterized by a group of congenital malformations such as: crytophthalmos, syndactyly, abnormal genitalia, and malformations of the nose, ears, and larynx. Although cryptophthalmos is considered as a main feature of Fraser syndrome, its absence does not exclude the diagnosis. Clinical diagnosis can be made by Thomas Criteria. Here we present the first documented case of Fraser Syndrome in Aleppo, Syria that is characterized by bilateral anophthalmia and intrahepatic biliary atresia.
Case presentation
During pregnancy, several ultrasound scans revealed hyperechoic lungs, ascites, and unremarkable right kidney at the 19th-week visit; bilateral syndactyly on both hands and feet at the 32nd-week visit. On the 39th week of gestation, the stillborn was delivered by cesarean section due to cephalopelvic disproportion. Gross examination showed bilateral anophthalmia, bilateral syndactyly on hands and feet, low set ears, and ambiguous genitalia. Microscopic examination of the lung, spleen, liver, ovary, and kidneys revealed abnormalities in these organs.
Conclusion
The diagnosis of Fraser syndrome can be made prenatally and postnatally; prenatally by ultrasound at 18 weeks of gestation and postnatally by clinical examination using Thomas criteria. Moreover, intrahepatic biliary atresia was not described previously with Fraser syndrome; this recommends a more detailed pathologic study for Fraser syndrome cases.
Similar content being viewed by others

Background
Fraser syndrome or cryptophthalmos syndrome [1], also known as Fraser-Francois syndrome, Meyer-Schwickerath’s syndrome, Ulrich-Feichtiger syndrome or cryptophthalmos-syndactyly syndrome [2, 3], is a rare autosomal recessive disease characterized by a group of congenital malformations, such as cryptophthalmos; syndactyly; abnormal genitalia; malformation of the nose, ears, and larynx; cleft lip; skeletal defects; umbilical hernia; mental retardation, and renal agenesis [1].
There are extremely few reported cases of Fraser syndrome with anophthalmia [4,5,6,7]; as Cryptophthalmos accounts for 88% of ocular malformations [8].
Genetically, mutations in FRAS1, FREM1, FREM2, and GRIP1 are responsible for the disease. These genes are essential for the regulation of epidermal-basement membrane adhesion and organogenesis during the embryonic period [9,10,11].
Fraser syndrome was first described by George Fraser in 1962 [6]. In which 25% of affected children are stillborns [12]. The estimated prevalence of Fraser syndrome is below 0.43 per 100,000 live-born infants and 11.06 per 100,000 stillbirths [13].
The diagnosis can be made prenatally by ultrasound, or clinically using Thomas criteria- two major criteria and one minor criterion, or one major criterion and four minor criteria are sufficient for diagnosis [1].. The major criteria are: cryptophthalmos, syndactyly, abnormal genitalia, and a sib with cryptophthalmos syndrome. The minor criteria are: cleft lip and/or palate, skeletal defects, and congenital malformation of the nose, ears, and larynx [1].
Prognosis of Fraser Syndrome depends on type and severity of the malformations and their possibility to be fixed [3, 14]..
Here we present the first documented case of Fraser syndrome in Aleppo, Syria that is characterized by bilateral anophthalmia and intrahepatic biliary atresia.
Case presentation
A 19-year-old woman (G2P1), married as a non-consanguineous marriage, with no past medical or surgical history, came to the outpatient prenatal clinic for routine visits.
Prenatal ultrasound scans of several visits revealed:
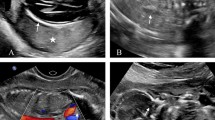
Hyperechoic lungs, ascites, unremarkable kidney (right), normal sucking reflux and a three-vessels umbilical cord in the 20th-week visit (Fig. 1).
Relative shortness in upper and lower limbs, nuchal edema, bilateral syndactyly on both hands and feet, hydrops fetalis, the abdominal circumference was 420 mm, enlargement of the lungs, and cardiac compression. The ultrasound estimated weight was 4000 g in the 25th-week visit (Fig. 2).

Ultrasound scans of the 20th-week visit: yellow arrow indicates liver, black arrow indicates lung, red arrow indicates kidney and blue arrow indicates ascites

Ultrasound scans of the 25th-week visit showed: relative shortness in upper and lower limbs, nuchal edema, bilateral syndactyly on both hands and feet, hydrops fetalis, enlargement of the lungs, and cardiac compression
After 39 weeks of gestation, the mother underwent a cesarean section for cephalopelvic disproportion. The fetus was stillborn due to respiratory insufficiency, weighing 3600 g. Gross examination showed multiple abnormalities including:
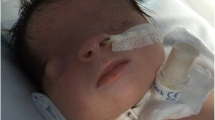
Bilateral anophthalmia, pseudo-hypertelorism, low-set ears, flat nasal bridge, bilateral syndactyly on hands and feet, cutaneous and subcutaneous edema, large-volume ascites, (Fig. 3 a) and ambiguous genitalia (Fig. 3 b).

Stillborn with bilateral anophthalmia, pseudo-hypertelorism, low-set ears, flat nasal bridge, bilateral syndactyly on hands and feet, cutaneous and subcutaneous edema, large-volume ascites (A) and ambiguous genitalia (B)
Autopsy revealed: normal airways, lung enlargement, no significant cardiac abnormalities, unilateral renal agenesis (right), a gonad and Mullerian structure were found on the left posterior pelvic wall, and the most outstanding find was the congested liver capsule which led us to do pathophysiologic study to find out the reason (Fig. 4).

Red arrows indicate right and left common iliac arteries, blue arrow indicates bladder, yellow arrow indicates the left ureter, green arrow indicates Mullerian duct and black arrow indicates gonad. The figure shows an absent right ureter and kidney
Microscopic examination of multiple sections of the lung, spleen, liver, ovary, and kidneys showed:
Dilated pleural lymphatic vessels (Fig. 5 a), interstitial fibroblast hypertrophy (Fig. 5 b) and vascular wall thickening in the lung (Fig. 5 c).
Congestion of red pulp with hemosiderin accumulation and immature white pulp of the spleen (Fig. 6).

Dilated pleural lymphatic vessels (A), interstitial fibroblast hypertrophy (B) and vascular wall thickening in the lung (C)

Congestion of red pulp with hemosiderin accumulation and immature white pulp of the spleen
Cholestasis with biliary atresia (Fig. 7 a, d, c) and biliary plugs (Fig. 7 B, C), portal spaces fibrosis (Fig. 7 a, d, e) with diffused congestion and diffused chronic inflammatory infiltrations with lymphocytes in the liver (Fig. 7 B).

Cholestasis with biliary atresia (A, D, C) and biliary plugs (B, C), portal spaces fibrosis (A, D, E) with diffused congestion and diffused chronic inflammatory infiltrations with lymphocytes in the liver (B)
While kidney sections revealed increased subcapsular nephrogenic structures (Fig. 8 a) and glomerular changes, such as decreasing subcapsular spaces (Fig. 8 b) and changing in the simple squamous epithelium of the outer parietal layer of the glomerular capsule into cuboidal epithelium (Fig. 8 c).

Subcapsular nephrogenic structures (A) and glomerular changes, such as decreasing subcapsular spaces (B) and changing in the simple squamous epithelium of the outer parietal layer of the glomerular capsule into cuboidal epithelium in the kidney (C)
As for the gonads, there were clusters of crowded immature primordial follicles (Fig. 9 a) and the Mullerian duct derivative was made up of epithelium resembling that of the Fallopian tube (Fig. 9 b).

Clusters of crowded immature primordial follicles (A) and the Mullerian duct derivative was made up of epithelium resembling that of the Fallopian tube (B)
Subsequently, based on microscopic examination of the lung and the presence of congenital malformations such as anophthalmia, bilateral syndactyly and renal agenesis, we suspected: plexiform pulmonary arteriopathy, Fraser Syndrome and Lenz microphthalmia syndrome as differential diagnosis for our case, but Fraser Syndrome was the most suitable one depending on Thomas et al. criteria.
The parents refused the genetic tests for the stillborn due to familial reasons.
Discussion and conclusion
Fraser Syndrome (OMIM 219000) is a rare autosomal recessive disease; probable mutations responsible for this disease are: FRAS1, FREM1, FREM2, and GRIP1 [9,10,11].
There are more than 250 reported cases of Fraser syndrome in medical literature [15].
Thomas et al. created the diagnostic criteria for Fraser syndrome [1]. The major criteria are: crytophthalmos, syndactyly, abnormal genitalia, and a sib with cryptophthalmos syndrome. The minor criteria are: cleft lip and/or palate, skeletal defects, and congenital malformation of the nose, ears, and larynx. Two major criteria and one minor criterion, or one major criterion and four minor criteria are sufficient for diagnosis [1].. alternatively, According to Slavotinek A et al. the diagnosis of Fraser syndrome can be made by the presence of one major criterion and one minor criterion [8].
In our case, the stillborn neonate had: bilateral anophthalmia, bilateral syndactyly (major) on hands and feet, ambiguous genitalia (major), low set ears (minor), unilateral renal agenesis (minor), and malformation of the nose (minor).
We have two major and three minor criteria that clinically affirm Fraser syndrome diagnosis according to Thomas and Slavotinek et al. criteria.
Cryptophthalmos; a continuous skin covering a normal or malformed eye [16], is the most common feature of Fraser Syndrome; it accounts for 88% of ocular malformations. However, other malformations can be present instead, such as microphthalmia; a small eye with normal eyelids [17], corneal opacification, and anophthalmia [8].
Anophthalmia; Inexistence of the eye [16], accounts for 6% of ocular malformations, and there are extremely few reported cases of Fraser syndrome with anophthalmia [4,5,6, 8].
Plexiform pulmonary arteriopathy and lenz microphthalmia syndrome were also differential diagnoses for our case, but the presence of congenital malformations led us to exclude plexiform pulmonary arteriopathy.
Lenz microphthalmia syndrome is characterized by anophthalmia or microphthalmia, in addition to mental retardation; external ear; digital, cardiac, skeletal and urogenital anomalies. It differs from Fraser syndrome by the presence of spinal and dental anomalies [18].
The definitive diagnosis of Fraser syndrome is only made by genetic analysis, detecting for FRAS1, FREM1, FREM2, and GRIP1 genes mutations, and this was a limitation in our case because the parents refused the genetic tests for the stillborn.
Prenatal diagnosis of Fraser syndrome is possible by ultrasound at 18 weeks of gestation with the following diagnostic signs:Hyperechogenic lungs, laryngeal stenosis/atresia, oligohydramnios, ascites, renal agenesis/dysplasia, microphthalmia/hypertelorism, hydrocephalus, syndactyly, ear defects and ambiguous genitals [19,20,21].
Prenatal diagnosis of Fraser syndrome provides a better management for infants with severe malformations in the respiratory tract [21].
In our case, prenatal ultrasound scans revealed: hyperechoic lungs, ascites, and an unremarkable right kidney at the 19th-week visit and bilateral syndactyly on both hands and feet at the 32nd-week visit.
Nevertheless, prenatal diagnosis is limited in relation to incidence of prenatal diagnosis obtained of Fraser syndrome. Berg et al. reported that prenatal diagnosis of FS was only in 43.75% (range 16–23 weeks of gestational age; sample size: 8) [19].
However, the rarity and the lack of knowledge of this syndrome in Syria made the diagnosis challenging. So that, the final probable diagnosis was made after birth.
Neonatal Cholestasis can occur due to deficiency in the excretion of bile, either for obstructive reasons or due to lack of bile secretion into the bile canaliculus [22].
Intrahepatic biliary atresia (paucity of intrahepatic bile ducts) can cause neonatal cholestasis [23]. It occurs in syndromic or non-syndromic form. The syndromic form, such as Alagille syndrome [24]. The non-syndromic form is associated with chromosomal disorders, metabolic or viral diseases, altered bile acid metabolism, and cystic fibrosis [25].
To sum up, intrahepatic biliary atresia was not described previously with Fraser Syndrome; this recommends a more detailed pathologic study for Fraser syndrome cases.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Thomas IT, Frias JL, Felix V, De Leon LS, Hernandez RA, Jones MC, Reynolds JF. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986 Sep;25(1):85–98.
Dumitru A, Costache M, Lazaroiu AM, Simion G, Secara D, Cirstoiu M, et al. Fraser syndrome - a case report and review of literature. Mædica. 2016;11(1):80–3.
Touré A, Diomandé IA, Nouraly H, Bérété R, Koffi KV, Kodjikian L. Bilateral cryptophthalmos in Fraser syndrome: case report and review of the literature. J Fr Ophtalmol. 2015;38(5):e97–100.
Pankau R, Partsch CJ, Jänig U, Meinecke R. Fraser (Cryptophthalmos-syndactyly) syndrome: a case with bilateral anophthalmia but presence of normal eyelids. Genetic counseling (Geneva, Switzerland). 1994;5(2):191–4.
Hafidi Z, Daoudi R. Bilateral anophthalmia in Fraser syndrome: report of a case. The Pan African medical journal. 2013;15:118-.
Mahadevan B, Bhat BV, Sastri AT, Rao S, Kusre G. Fraser syndrome with unusual features. A case report. J Anat Soc India. 2002;51(1):59–60.
Mbonda A, Endomba FT, Kanmounye US, Nkeck JR, Tochie JN. Diagnosis of Fraser syndrome missed out until the age of six months old in a lowresource setting: a case report. BMC Pediatr. 2019;19:292.
Slavotinek A, Tifft C. Fraser syndrome and cryptophthalmos: review of thediagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. J Med Genet. 2002;39(9):623–33.
Sag SO, Gorukmez O, Ture M, Sahinturk S, Topak A, Gulten T, Schanze D, Yakut T, Zenker M. A novel mutation in the FRAS1 gene in a patient with Fraser syndrome. Genet Couns. 2015;26(1):21.
Schanze D, Kayserili H, Satkın BN, Altunoglu U, Zenker M. Fraser syndrome due to mutations in GRIP1--clinical phenotype in two families and expansion of the mutation spectrum. American journal of medical genetics. Part A. 2013 Dec 19;164(3):837–40.
Short K, Wiradjaja F, Smyth I. Let's stick together: the role of the Fras1 and Frem proteins in epidermal adhesion. IUBMB Life. 2007;59(7):427–35.
Fraser GR. Our genetical ‘load’. A review of some aspects of genetical variation. Ann Hum Genet. 1962 May;25(4):387–415.
Martinez-Frias ML, Bermejo ES, Felix V, Calvo RC, Ayala AG, Hernández FR. Fraser syndrome: frequency in our environment and clinical-epidemiological aspects of a consecutive series of cases. Anales espanoles de pediatria. 1998 Jun;48(6):634–8.
Barisic I, Odak L, Loane M, Garne E, Wellesley D, Calzolari E, et al. Fraser syndrome: epidemiological study in a European population. Am J Med Genet A. 2013;161A;(5):1012–8.
Van Haelst MM, Scambler PJ. Fraser syndrome collaboration group, Hennekam RC. Fraser syndrome: a clinical study of 59 cases and evaluation of diagnostic criteria. Am J Med Genet A. 2007 Dec 15;143(24):3194–203.
Kanhere S, Phadke V, Mathew A, Irani SF. Cryptophthalmos. The Indian Journal of Pediatrics. 1999 Sep 1;66(5):805–8.
Verma AS, FitzPatrick DR. Anophthalmia and microphthalmia. Orphanet journal of rare diseases. 2007 Dec 1;2(1):47.
Forrester S, Kovach MJ, Reynolds NM, Urban R, Kimonis V. Manifestations in four males with and an obligate carrier of the Lenz microphthalmia syndrome. Am J Med Genet. 2001 Jan 1;98(1):92–100.
Berg C, Geipel A, Germer U, Pertersen-Hansen A, Koch-DÖrfler M, Gembruch U. Prenatal detection of Fraser syndrome without cryptophthalmos: case report and review of the literature. Ultrasound in Obstetrics and Gynecology: The Official Journal of the International Society of Ultrasound in Obstetrics and Gynecology. 2001 Jul;18(1):76–80.
Tessier A, Sarreau M, Pelluard F, André G, Blesson S, Bucourt M, et al. Fraser syndrome: features suggestive of prenatal diagnosis in a review of 38 cases. Prenat Diagn. 2016;36(13):1270–5.
De Bernardo G, Giordano M, Di Toro A, Sordino D, De Brasi D. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Dec 1;41(1):86.
Khalaf R, Phen C, Karjoo S, Wilsey M. Cholestasis beyond the neonatal and infancy periods. Pediatric gastroenterology, hepatology & nutrition. 2016 Mar 1;19(1):1–1.
Suchy FJ. Clinical problems with developmental anomalies of the biliary tract. InSeminars in gastrointestinal disease 2003 Oct (Vol. 14, No. 4, pp. 156–164).
Arora K. Intrahepatic biliary atresia. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverintrahepaticbiliaryatresia.html. Accessed November 23rd, 2019.
Koçak N, Gürakan F, Yüce A, Çaglar M, Kale G, Gögüs S. Nonsyndromic paucity of interlobular bile ducts: clinical and laboratory findings of 10 cases. J Pediatr Gastroenterol Nutr. 1997 Jan 1;24(1):44–8.
Acknowledgements
Not applicable.
Funding
There are no sources of funding.
Author information
Authors and Affiliations
Contributions
HN: Supervisor, obstetrician, performed autopsy. SA: Supervisor, Pathologist. MZBA, HS, RB, MNK and AA: analyzed and interpreted the patient data, Wrote the manuscript, revision. MNK was the corresponding author. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not required for case reports at our hospital. Single case reports are exempt from ethical approval in our institution.
Consent for publication
The mother provided written informed consent for the publication of potentially identifying images clinical and details, on behalf of herself and the child.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Brimo Alsaman, M.Z., Agha, S., Sallah, H. et al. Bilateral anophthalmia and intrahepatic biliary atresia, two unusual components of Fraser syndrome: a case report. BMC Pregnancy Childbirth 20, 358 (2020). https://doi.org/10.1186/s12884-020-03048-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12884-020-03048-x