
Abstract
Background
Recent studies suggest that there is a link between the gut microbiota and glucose metabolism. This study aimed to compare the gut microbiota during early pregnancy of women with hyperglycymia to those with normal blood glucose.
Methods
Gut microbial composition was analysed in 22 women with hyperglycaemia and 28 age-matched healthy controls during their first prenatal visits (< 20 weeks) using high throughput sequencing of the V3-V4 region of the 16S ribosomal RNA gene. Hyperglycemia was diagnosed based on the criteria recommended by the International Association of Diabetes and Pregnancy Study Groups in 2010.
Results
Women with hyperglycemia in pregnancy (HIP) had significantly lower microbial richness and diversity compared with healthy pregnant women. The proportions of the Firmicutes and Bacteroidetes phyla and the ratio of Firmicutes:Bacteroidetes were not different between the two groups. We observed that individuals with HIP had an increased abundance of Nocardiaceae, Fusobacteriaceae, etc., whereas healthy controls had significantly higher levels of Christensenellaceae, Clostridiales_vadinBB60_group, Coriobacteriaceae, etc. Similarly, levels of the members of the Ruminococcaceae family, including Ruminococcaceae_UCG-014, Ruminococcaceae_UCG-003, and Ruminococcaceae_UCG-002, were significantly reduced in the HIP group and were negatively correlated with HbA1c. HbA1c levels were positively correlated with Bacteroidaceae and Enterobacteriaceae and negatively correlated with Christensenellaceae, etc. CRP was positively correlated with the Bacteroidaceae and Fusobacteriaceae families and the Fusobacterium genus.
Conclusions
Our study revealed that individuals with HIP have gut microbial dysbiosis and that certain bacterial groups are associated with glucose metabolism during pregnancy. Further study is needed to provide new ideas to control glucose by modifying the gut microbiota.
Similar content being viewed by others

Background
Hyperglycaemia in pregnancy (HIP) is a very common medical disorder during pregnancy that consists of two categories: pregestational diabetes (PGDM), antedating pregnancy, and gestational diabetes mellitus (GDM), initially diagnosed during gestation [1]. The increasing prevalence of HIP and its related adverse pregnancy outcomes are a global health concern [2,3,4]. Dysbiosis in the human gut may be a vital risk factor for abnormal glucose metabolism and was first discovered by Backhed F and his colleagues in 2004 who found that conventionalisation of germ-free mice with microbiota from conventionally raised animals produced a marked increase in body fat content and insulin resistance despite decreased food intake [5]. Since then, the association between the gut microbiota and glucose metabolism has been a popular research topic.
Data from human and animal studies suggest that individuals with obesity and insulin resistance have a lower bacterial diversity [6, 7]. A lower prevalence of the Bacteroidetes phylum and a higher prevalence of the Firmicutes phylum have been shown to be associated with obesity [8, 9]. Obesity was linked to changes in the Bacteroidetes/Firmicutes ratio, which has been associated with an increased capacity to harvest energy from the diet [9]. Prevotella copri and Bacteroides vulgatus are the main species associated with the biosynthesis of branched-chain amino acids, insulin resistance, and glucose intolerance [10]. Furthermore, the levels of butyrate-producing bacteria were reduced in diabetic individuals [10], and butyrate supplementation in obese, prediabetic mice significantly improved insulin resistance, hyperinsulinaemia, and hyperglycaemia [11].
While the majority of previous studies have been concerned with the associations between the gut microbiota and obesity or type 2 diabetes (T2DM) [8,9,10, 12, 13], some recent studies have explored changes in the microbiota during pregnancy. Decreased insulin sensitivity and enhanced nutrient absorption are beneficial for normal pregnancy, by supporting foetal growth and meeting the energetic demands of lactation [14]. Hormones, immunity and metabolism change substantially in pregnancy. However, human microbiota studies have also demonstrated that the gut microbiota changes dramatically during pregnancy [15]. From the first to third trimesters, Bifidobacteria, Proteobacteria, and lactic-acid-producing bacteria increase and butyrate-producing bacteria decrease [15].
What is the relationship between the gut microbiota and glucose metabolism disorders in the context of pregnancy? A study based on whole-metagenome shotgun sequencing discovered that the composition of the gut microbiota was different between controls and women with GDM at 21–29 weeks. GDM patients showed an increase in Parabacteroides distasonis and Klebsiella variicola, whereas controls showed an enrichment of Methanobrevibacter smithii, Alistipes spp., Bifidobacterium spp., and Eubacterium spp. [16]. Another recent study by Fangqing Zhao also demonstrated that GDM patients had lower microbial richness and diversity than controls within 1–2 days before delivery [17]. To date, there is no information available on the gut microbiota in relation to glucose metabolism disorder in early pregnancy.
In this study, we used 16S rRNA gene amplicon sequencing to analyse the gut microbiota in women with HIP and healthy controls during early pregnancy. We also explored the connections between gut microbiota composition and glucose metabolism-related indicators. The objective was to further understand the associations between the gut microbiota and HIP in early pregnancy.
Methods
Study population
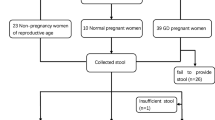
In this study, 50 pregnant women in early pregnancy (< 20 weeks gestation) were recruited during their first prenatal visits at the Maternity and Child Health Center of Suzhou Municipal Hospital between 1 Nov 2015 and 31 Oct 2017. All participants were Han Chinese. Twenty-two pregnant women had different degrees of glucose metabolism disorders were signed into the HIP group. Among HIP group, 8 had type 2 diabetes mellitus before pregnancy who were only through diet/lifestyle to control glucose. Based on the diagnostic criteria recommended by the International Association of Diabetes and Pregnancy Study Groups (IADPSG) in 2010 [18], 5 overt diabetes and 9 GDM were diagnosed by hemoglobin A1c (HbA1c) or fasting glucose (FBG) and confirmed by an oral glucose tolerance test (OGTT). Twenty-eight pregnant women with fasting plasma glucose lower than 5.1 mmol/L were assigned into control group. Additionally, they had a normal glucose tolerance during 24–26 weeks’ gestation to comfirm they were healthy Patients were excluded if they met one of the following criteria: 1) history of antibiotic therapy within the last 3 months; 2) chronic diseases requiring medication; and 3) history of smoking. Written informed consent was obtained from all subjects before their participation. Ethics approval was obtained from the hospital.
Characteristic data collection
The anthropometric data (pre-pregnancy weight (pre-weight), height, pre-body mass index (pre-BMI), waist circumference (waist) and hip circumference (hip)), lifestyle factors and history of medication and treatments were obtained from clinical medical records. Pre-BMI was calculated as the subject’s pre-weight (kg) divided by their height squared (m2). Waist and hip were measured in an erect position at the narrowest point between the iliac crest and the lower costal margin and at the level of the pubic symphysis, respectively. Systolic and diastolic blood pressure was measured twice (YuTu Model XJ11D, YuTu Company, Shanghai, China) with the participant in a sitting position, and the mean value was used for further analysis. The subjects were given instructions by a physician to how to be fasting prior to the collection of samples. After 8 h of fasting, venous blood samples were collected between 07:00 and 09:00 for the assessment of metabolic biomarkers (FBG, TC, TG, LDL-C, HDL-C, etc) using a fully automatic biochemical analyser (Hitachi 7000, Tokyo, Japan). HbA1c level was detected by using high-performance liquid chromatography (HLC-723G8, Tosoh Bioscience, Japan) Insulin content was measured using a Human Insulin ELISA Kit (Sigma-Aldrich, St. Louis, MO, USA).
Sample collection, DNA extraction and 16S rRNA PCR
Fresh stool samples were collected from pregnant women at home using sterile faecal collection tubes, and samples were immediately transferred to the lab within 6 h and then stored at − 80 °C under a uniform protocol before DNA extraction. Total microbial genomic DNA from 250 mg stool was extracted using the Qiagen QIAamp DNA Stool Mini Kit (Qiagen, Shanghai, China) following the manufacturer’s protocols. A NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA) and 1% agarose gel were employed to evaluate the purification and quality of the DNA samples. The V3-V4 hypervariable regions of the bacterial 16S rRNA gene were amplified using 338F and 806R primers on a thermocycler PCR system (GeneAmp 9700, ABI, USA). PCR reactions (20 μL: 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 1.6 μL of 5 μM primers, 0.4 μL of FastPfu Polymerase and 10 ng of template DNA) were performed according to the following cycling programme: denaturation at 95 °C for 3 min; followed by 27 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C 45 s; and 72 °C for 10 min. Each sample was analysed in triplicate. PCR products with clear electrophoresis strips and an expected size were extracted from 2% agarose gel, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using QuantiFluor™-ST (Promega, Madison, WI, USA).
Illumina MiSeq sequencing and data processing
After purification and equivalent mixing of the PCR products, paired-end (2 × 300 bp) Illumina MiSeq (Illumina, San Diego, USA) sequencing was carried out at Nanjing Decode Genomics BioTech Co., Ltd. (Nanjing, China) according to the standard protocols. We trimmed and filtered the reads (FastQ files) by using Trimmomatic [19] and merged the paired reads with FLASH [20]. Operational taxonomic units (OTUs) were clustered using USEARCH [20, 21](version 7.1 http://drive5.com/uparse/) at the 97% similarity level. UCHIME was performed to filter out the chimeric sequences [22]. Taxonomic classification of OTUs was performed using the RDP Classifier algorithm [23] (http://rdp.cme.msu.edu/) by aligning representative sequences to the Silva (SSU123) 16S rRNA database [24].
Statistical analysis
Continuous data are expressed as the median (interquartile range). Comparisons between groups were performed using Mann-Whitney U tests or Fisher’s exact test with adjusted age. The alpha diversity (including the Chao1 index and Shannon diversity index) of bacteria within individuals was used to evaluate the number of different taxa, species richness and evenness of bacterial taxa. Microbial community composition differences (beta diversity) between groups were analysed using permutational multivariate analysis of variance (PMANOVA) of unweighted or weighted UniFrac distances (Bray-Curtis distance method, 999 permutations), and the results are illustrated by principal coordinates analysis (PCoA). Canonical correspondence analysis (CCA) was used to assess the impact of each of the factors listed on microbial abundance. Differences in microbial abundance (at the phylum, family, genus and species levels) were analysed using the Mann-Whitney U test. P-values were adjusted for false discovery rate (FDR) using the Benjamini and Hochberg method. P < 0.05 was considered statistically significant. Bacterial community diversity and composition were illustrated using Boxplot graphs and cluster and heatmap diagrams in R (ggplot2 package). Statistical analyses were performed using Graph Pad Prism 6 software (GraphPad, San Diego, USA) or SPSS 19 software (IBM, New York, USA).
Results
Clinical characteristics and biochemical variables
The clinical characteristics of the 50 pregnant women at < 20 weeks gestation are presented in Table 1. Twenty-eight controls and 22 women with HIP were matched for maternal age. Fasting serum levels of seven biochemical variables (fasting blood glucose (GLU), triglycerides (TGs), high-density lipoprotein (HDL), low-density lipoprotein (LDL), HbA1c and C-reactive protein (CRP)) were measured in all participants. There were significant differences in GLU, TGs, HDL, LDL, HbA1c and CRP between the two groups, while TC did not show significant differences (Table 1), with subjects in the HIP group having a more disturbed metabolic profile.
Bacterial community structure (beta diversity) in the two study groups
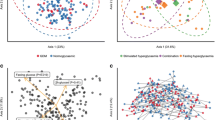
A total of 912 OTUs (with 97% similarity) were clustered in this study. The PMANOVA results showed significant differences between the HIP and control groups (P = 0.001). However, we also found that the samples from 10 pregnant women (cluster 2) showed a bacterial composition distinct from those of the rest of the study cohort (cluster 1) (pMANOVA P = 0.001, Fig. 1). There were no significant differences in anthropometric or biochemical variables between cluster 1 and cluster 2 (Table S1). Based on microbial analysis, it was found that the abundance of the Bacteroidaceae family was higher in Cluster 1 (32.1 [18.2–45.8] vs. 7.6 [0.9–14.2] %, P < 0.0001), whereas the proportion of the Prevotellaceae family was more abundant in Cluster 2 (2.6 [0–7.9] vs. 38.5 [17.0–59.9] %, P < 0.0001). Consistent with previous enterotype structure reports [25], Cluster 1 and Cluster 2 were classified as Bacteroides and Prevotella enterotypes. However, no significant relationship was found between enterotype and disease status (P = 0.103, Fisher’s exact test). Furthermore, we examined the principal components with a contribution greater than 4% and found that the first and second principal components were not only significantly correlated with enterotype (P < 0.0001, Mann–Whitney U test) but were also significantly correlated with disease status (P < 0.05, Mann–Whitney U test). These results suggested that in addition to enterotype, disease status was also an important factor affecting the gut microbial composition in our study (Table S2).

PCoA of the bacterial community composition based on Bray Curtis analysis. Dots represent individuals (women with HIP = red, controls = blue). Cluster 1 and Cluster 2 are represented as Bacteroides and Prevotella enterotypes, respectively
The microbiotas of women with HIP and healthy pregnant women are different in early pregnancy
We compared the gut microbial composition between 22 HIP and 28 healthy women in early pregnancy. We found that the HIP group showed significantly lower microbial richness and α-diversity than the healthy controls (P < 0.05 for both indexes, Mann–Whitney U test; Fig. 2, Fig. S1). PMANOVA analysis showed that there was a significant distance between the HIP and control groups (P < 0.001). Consistent with the PMANOVA results, CCA also showed a marked separation of women with HIP from the controls (Fig. S2).

Decreased gut microbial richness and diversity in women with HIP
Richness and α-diversity (Chao1 and Shannon index) of the two cohorts at the OTU level. Boxplots showing both richness and diversity values. *P < 0.05; **P < 0.01; ***P < 0.001.
At the phylum level, Firmicutes and Bacteroidetes were the dominant phyla, accounting for ~ 91.5% of all sequences in individuals, whereas 0.04% was accounted for by unknown bacteria (Table S3). The ratio of Firmicutes to Bacteroidetes was similar in patients and controls (1.33 vs. 1.27, P = 0.44). Other less-prominent phyla including Saccharibacteria and Fusobacteria were enriched in the HIP group, while Lentisphaerae and Tenericutes were enriched in the healthy controls (P < 0.05, Fig. 3). The Fusobacterial class and Fusobacteriales order were also enriched in the HIP group.

Phylum-level differences in bacteria in the faecal samples of controls and women with HIP. Boxplot shows the phyla that differed significantly between the women with HIP (red) and healthy controls (blue). The percentage of the phylum is indicated on the y axis. Boxplots showing the 25th and 75th percentiles with a line at the median
At the family level, women with HIP had a significantly higher proportion of Nocardiaceae, Saccharibacteria_norank, and Fusobacteriaceae, while the proportions of Christensenellaceae, Clostridiales_vadinBB60_group, Mollicutes_RF9_norank, Oxalobacteraceae, Victivallaceae, Rhodospirillaceae, and Coriobacteriaceae were higher in healthy controls (P < 0.05; Fig. 4). In addition, many genera showed significantly lower abundances in women with HIP than in controls, and these genera included Ruminococcaceae_UCG-014, Christensenellaceae_R-7_group, Ruminococcaceae_UCG-003, and Ruminococcaceae_UCG-002. Differences at the species level were also found between women with HIP and healthy controls and were consistent with the findings at the genus level (Table S3). These findings suggested that women with HIP had gut microbial dysbiosis.

Families that differed significantly between women with HIP and healthy controls. The relative abundance of the bacterial families was significantly different between women with HIP (red) and healthy pregnant women (blue) (top 10). The percentage of microbiota is indicated on the y axis. Boxplots showing the 25th and 75th percentile with a line at the median
Correlations between the gut microbiota and clinical characteristics
To assess associations between clinical characteristics and the gut microbiota, Spearman’s correlation coefficient was calculated. At the family level, pre-BMI, pre-weight and waist were significantly negatively correlated with the Clostridiales_vadinBB60_group (Fig. 5a). pre-BMI was also negatively associated with Ruminococcaceae_UCG-005 and Ruminococcaceae_UCG-014. In addition, waist was negatively correlated with the Gastranaerophilales_norank and positively correlated with Erysipelotrichaceae.

Correlation of clinical characteristics with microbial abundance
Correlation analysis results of all samples at the family level (a) and the genus level (b). Spearman’s rank correlation coefficients and P-values for the correlations are shown. *P < 0.05; **P < 0.01; ***P < 0.001
The relationship between gut microbial composition and blood glucose metabolism was also assessed. HbA1c was positively correlated with the relative abundance of Proteobacteria, Fusobacteria, and Saccharibacteria and negatively correlated with Lentisphaerae (P < 0.05). Furthermore, HbA1c was positively associated with the abundance of the Bacteroidaceae family from the Bacteroidetes phylum (Fig. 5a). A positive correlation between HbA1c, GLU and the Enterobacteriaceae family was observed (P < 0.05) (Fig. 6a). A higher HbA1c level was correlated with a lower abundance of multiple families, such as Ruminococcaceae, Christensenellaceae, Victivallaceae, Rhodospirillaceae, and Micrococcaceae, while GLU exhibited a similar trend (Fig. 5 a, Fig. 6 b, C). At the genus level, Bacteroides, Bilophila and Lachnospiraceae_uncultured were positively correlated with HbA1c, while several genera of the Ruminococcaceae family were negatively correlated with HbA1c and GLU (Fig. 5 b).

Bacterial abundance and its associations with HbA1c and CRP profile a. Positive correlation between HbA1c concentration and the Enterobacteriaceae family. b, c. Negative correlation between the Ruminococcaceae and Christensenllaceae families with HbA1c. d: Positive correlation between the Fusobacterium genus and CRP concentration
Low-grade chronic inflammation plays a key role in the pathophysiology of glucose disorders. CRP is mainly used as a marker of inflammation and has been proven to be associated with diabetes. In our study, CRP was positively correlated with the relative abundance of Proteobacteria, Fusobacteria, and Saccharibacteria and negatively correlated with Lentisphaerae (P < 0.05). Fusobacteriaceae was the main family of the Fusobacteria phyla. Further analysis within Fusobacteria indicated that the Fusobacteriaceae family and the Fusobacterium genus were also positively associated with CRP (P < 0.05) (Fig. 5a, Fig. 6d). We also found that the abundance of the Bacteroidaceae family from the Bacteroidetes phylum was positively associated with CRP.
Discussion
This study is the first to our knowledge to compare the stool microbiota of patients who have hyperglycaemia (HIP) with that of controls in early pregnancy. We discovered that the participants were separated into two clusters. This finding was not explained by differences in anthropometric or biochemical variables, and the two clusters corresponded to Bacteroides and Prevotella enterotypes, respectively, in accordance with the recently-proposed theory of enterotypes [26, 27]. No significant correlation between enterotype and disease status was found in our study. However, we should fully take enterotype into account in gut microbiota research, although our current study lacks the sample size to make these conclusions. Furthermore, by examining the top six principal components, we found that the first and second principal components were also significantly correlated with disease status. We concluded that glucose metabolism disorders, in addition to enterotype, was a determining factor in explaining the gut microbial differences in our research.
We observed reduced richness and diversity in the faecal microbiota of the HIP group compared to that of the controls. Similar findings were reported for a variety of chronic diseases, including obesity [6, 7], inflammatory bowel disease [28], cancer [29] and even mental disorders [30], indicating that a decline in richness and diversity is a common characteristic in many kinds of diseases.
Most studies discovered that obesity is characterized by an increase in the Firmicutes phylum and a relative decrease in the Bacteroidetes phylum [7,8,9]. However, this phenomenon was quite contradictory in the context of type 2 diabetes. A study from Nadja Larsen and colleagues that included 18 male type 2 diabetes patients and 18 healthy male controls demonstrated that the relative abundance of Firmicutes was significantly reduced in individuals with type 2 diabetes [12]. Another study from China that included 71 type 2 diabetes patients and 74 controls also obtained similar results [24, 26]. However, the abundance of Firmicutes and Bacteroidetes and the ratio of Firmicutes:Bacteroidetes were similar between the two groups in our study. Our findings were consistent with previous research on polycystic ovary syndrome (PCOS) [31], GDM in the second trimester and GDM within 1–2 days before delivery [16, 17]. We speculate that the opposite results may be because the subjects in the latter four studies were all women who were much younger than those included in the first two studies. When comparing less-prominent phyla, we observed that Lentisphaerae and Tenericutes were enriched in healthy controls. Consistent with other studies, Tenericutes was more abundant in controls than in individuals with PCOS [31] or metabolic syndrome [32]. Therefore, we have reason to believe that bacteria from Tenericutes seem to play a protective role against several kinds of diseases.
The Clostridiales order, which can produce butyrate, was reduced in individuals with obesity [33], T2DM [24, 26] and gestational diabetes mellitus in the second trimester [16]. Our data supported this finding by showing that Clostridiales was enriched in healthy pregnant women in early pregnancy and that the Clostridiales_vadinBB60_group family was negatively associated with several obesity-related markers. Previous studies demonstrated that butyrate seemed to possess beneficial effects in terms of insulin sensitivity and energy balance [34]. The reason may partly explain why Clostridiales was more abundant in healthy subjects than in individuals with metabolic disease both in pregnancy and non-pregnancy conditions. The Christensenellaceae family, which has recently been reported in relation to leanness and healthy metabolism, was found in a significantly higher level in controls than in women with HIP and was negatively correlated with HbA1c in our study [35]. Previous studies have found that Christensenellaceae is negatively associated with the branch-chain amino acids (BCAAs) isoleucine and valine [34, 36]. BCAAs are positively associated with insulin resistance [37, 38]. Moreover, increased concentrations of three BCAAs (valine, leucine, isoleucine) and two aromatic amino acids (phenylalanine, tyrosine) were associated with future diabetes in the Framingham Heart Study [39]. All these results suggest that Christensenellaceae played a protective role against diabetes through altered amino acid metabolism.
We also observed that several genera from the Ruminococcaceae family were more abundant in controls than in women with HIP and were negatively correlated with HbA1c. Consistent with previous findings, Ruminococcaceae were depleted in women with GDM in the third trimester of pregnancy [40], in non-pregnant individuals with type 1 diabetes [41] and even in pre-diabetic subjects [36]. However, the opposite result was obtained in some other studies. Luisa F. Gomez-Arango and colleagues demonstrated that a high abundance of Ruminococcaceae may be related to adverse metabolic health in early pregnancy in overweight and obese women [42]. This finding was further supported by Kati Mokkala, who studied overweight and obese women in early pregnancy [43]. Ruminococcaceae is considered to produce short-chain fatty acids that have positive effects on metabolism [44]. The efficiency of Ruminococcaceae in the last two studies, which focused on overweight and obese women, may have been lower.
Enterobacteriaceae is a large family of Gram-negative bacteria that includes many of the familiar pathogens. A positive correlation of the Enterobacteriaceae family with HbA1c was observed in our study. Enterobacteriaceae was also found in a higher relative abundance in GDM patients in the second trimester [16] and in non-pregnant individuals with T2DM than in controls [45] and in individuals with some chronic diseases, such as colitis [46] and inflammatory bowel disease (IBD) [47]. A previous study showed that Enterobacteriaceae indicated a specific status of gut inflammation and colitis due to its role in inducing intestinal inflammation [46]. The level of the endotoxin-producing Enterobacter decreased in a morbidly obese human who lost 51.4 kg after 23 weeks of dietary intervention [48]. These results suggest that abnormalities of bacteria from the Enterobacteriaceae family may be associated with HIP.
The strengths of our research are that the research sample was composed of young adults with few concomitant medications and comorbidities. The main limitations of our present study are the small sample size and the fact that lifestyle and diet, which may affect both blood glucose levels and the gut microbiota, were impossible to assess in the present study.
In conclusion, our study suggests that women with HIP have dysbiosis of the gut microbiota. In particular, our results support the idea that certain members of the microbiota were associated with glucose metabolism during pregnancy. The gut microbiota may be a potential biomarker for patients with glucose metabolism disorder in early pregnancy. Future studies are needed to evaluate the diagnostic value of relevant microbial markers.
Availability of data and materials
The datasets used and analysed in the current study are available from the corresponding author upon reasonable request.
Abbreviations
- HIP:
-
Hyperglycaemia in pregnancy
- PGDM:
-
Pregestational diabetes
- GDM:
-
Gestational diabetes mellitus
- OGTT:
-
Oral glucose tolerance test
- T2DM:
-
Type 2 diabetes
- FLASH:
-
Fast length adjustment of short reads
- RDP:
-
Ribosomal Database project
- PMANOVA:
-
Permutational multivariate analysis of variance
- CCA:
-
Canonical correspondence analysis
- OUTs:
-
Operational taxonomic units
- PCOS:
-
Polycystic ovary syndrome
- BCAAs:
-
Branch-chain amino acids
- IBD:
-
Inflammatory bowel disease.
References
Cosson E, Carbillon L, Valensi P. High fasting plasma glucose during early pregnancy: a review about early gestational diabetes mellitus. J Diabetes Res. 2017;2017.
Association AD. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes—2018. Diabetes Care. 2018;41(Supplement 1):S13–27.
Lawrence JM, Contreras R, Chen W, Sacks DA. Trends in the prevalence of pre-existing diabetes and gestational diabetes mellitus among a racially/ethnically diverse population of pregnant women, 1999-2005. Diabetes Care. 2008;31(5):899–904.
Dunne F, Brydon P, Smith K, Gee H. Pregnancy in women with type 2 diabetes: 12 years outcome data 1990–2002. Diabetic Med. 2003;20(9):734–8.
Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101(44):15718–23.
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541.
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102(31):11070–5.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027.
Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535(7612):376.
Matheus V, Monteiro L, Oliveira R, Maschio D, Collares-Buzato C. Butyrate reduces high-fat diet-induced metabolic alterations, hepatic steatosis and pancreatic beta cell and intestinal barrier dysfunctions in prediabetic mice. Exp Biol Med. 2017;242(12):1214–26.
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085.
Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99.
Lain KY, Catalano PM. Metabolic changes in pregnancy. Clin Obstet Gynaecol. 2007;50(4):938–48.
Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150(3):470–80.
Kuang Y-S, Lu J-H, Li S-H, Li J-H, Yuan M-Y, He J-R, et al. Connections between the human gut microbiome and gestational diabetes mellitus. Gigascience. 2017;6(8):1–12.
J Wang, J Zheng , W Shi, N Du, X Xu, Y Zhang, et al. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut. 2018;67(9):gutjnl-2018-315988.
International Association of Diabetes and Pregnancy Study Groups Consensus Panel, Metzger BE, Gabbe SG, Persson B, Buchanan TA, Catalano PA, et al. International Association of Diabetes and Pregnancy Study Groups Recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33(3):676–82.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1.
Edgar R. UCHIME2: improved chimera prediction for amplicon sequencing. Biorxiv. 2016;074252.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41(D1):D590–D6.
Costea PI, Hildebrand F, Manimozhiyan A, Bäckhed F, Blaser MJ, Bushman FD, et al. Enterotypes in the landscape of gut microbial community composition. Nat Microbiol. 2018;3(1):8.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55.
Fugmann M, Breier M, Rottenkolber M, Banning F, Ferrari U, Sacco V, et al. The stool microbiota of insulin resistant women with recent gestational diabetes, a high risk group for type 2 diabetes. Sci Rep. 2015;5:13212.
Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55(2):205–11.
Chen J, Domingue JC, Sears CL. Microbiota dysbiosis in select human cancers: evidence of association and causality. Semin Immunol. 2017;32:25–34.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–63.
Lindheim L, Bashir M, Münzker J, Trummer C, Zachhuber V, Leber B, et al. Alterations in gut microbiome composition and barrier function are associated with reproductive and metabolic defects in women with polycystic ovary syndrome (PCOS): a pilot study. PLoS One. 2017;12(1):e0168390.
Lim MY, You HJ, Yoon HS, Kwon B, Lee JY, Lee S, et al. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut. 2016;gutjnl-2015-311326.
Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med. 2017;23(7):859.
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–17.
Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–99.
Org E, Blum Y, Kasela S, Mehrabian M, Kuusisto J, Kangas AJ, et al. Relationships between gut microbiota, plasma metabolites, and metabolic syndrome traits in the METSIM cohort. Genome Biol. 2017;18(1):70.
Stančáková A, Civelek M, Saleem NK, Soininen P, Kangas AJ, Cederberg H, et al. Hyperglycemia and a common variant of GCKR are associated with the levels of eight amino acids in 9,369 Finnish men. Diabetes. 2012; DB_111378.
Tai E, Tan M, Stevens R, Low Y, Muehlbauer M, Goh D, et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia. 2010;53(4):757–67.
Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17(4):448.
Crusell MKW, Hansen TH, Nielsen T, Allin KH, Rühlemann MC, Damm P, et al. Gestational diabetes is associated with change in the gut microbiota composition in third trimester of pregnancy and postpartum. Microbiome. 2018;6(1):89.
Huang Y, Li S-C, Hu J, Ruan H-B, Guo H-M, Zhang H-H, et al. Gut microbiota profiling in Han Chinese with type 1 diabetes. Diabetes Res Clin Pract. 2018;141:256–63.
Gomez-Arango LF, Barrett HL, McIntyre HD, Callaway LK, Morrison M, Nitert MD, et al. Connections between the gut microbiome and metabolic hormones in early pregnancy in overweight and obese women. Diabetes. 2016:db160278.
Mokkala K, Houttu N, Vahlberg T, Munukka E, Rönnemaa T, Laitinen K. Gut microbiota aberrations precede diagnosis of gestational diabetes mellitus. Acta Diabetol. 2017;54(12):1147–9.
Bearson SMD, Allen HK, Bearson BL, Looft T, Brunelle BW, Kich JD, et al. Stanton TB: profiling the gastrointestinal microbiota in response to Salmonella : low versus high Salmonella shedding in the natural porcine host. Infect Genet Evol. 2013;16:330–4.
Salamon D, Sroka-Oleksiak A, Kapusta P, Szopa M, Mrozińska S, Ludwig-Słomczyńska AH, et al. Characteristics of gut microbiota in adult patients with type 1 and type 2 diabetes based on next-generation sequencing of the 16S rRNA gene fragment. Pol Arch Med Wewn. 2018;128(6):336–43.
Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8(3):292–300.
Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79.
Fei N, Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013;7(4):880.
Acknowledgements
We thank Shuxiang Li for her assistance in collecting the stool samples (Clinical laboratory, the Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital).
Funding
This work was supported by research grants from the Science and Technology Development Plan Project of Suzhou (SYS201570 and SS201872), and Fund for Digestive Diseases and Nutrition Research Key Laboratory of Suzhou (SZS201620). The funding bodies had no role in the design of the study or collection, analysis, or interpretation of data. The writing of the manuscript was conducted strictly by the authors, unaffected by the funding bodies. The fee of analysing the gut microbiota using 16S rRNA gene amplicon sequencing was supported by the funder (SYS201570). The open access publication fee was paid by the funder (SS201872).
Author information
Authors and Affiliations
Contributions
BG, MZ and LC participated in the design of the study. MZ, QS, YW and SJ collected the samples. MC, YW and BG performed the statistical analysis. LC helped to interpret the results. All authors drafted, read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was approved by the Research Ethics Committees of the Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital (K20150010). Written informed consent was obtained from all the participants prior to enrolment in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1
Characteristics of Cluster 1 and Cluster 2. Table S2 PCoA results of OTUs profiles. Figure S1. Richness and a-diversity (ACE and Simpson index) of the two cohorts at the OTU level. Figure S2. Comparison of the microbiota community composition between HIPs and healthy pregnant women
Additional file 2 Table S3
. Differential microbial abundance between HIPs and controls
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, B., Zhong, M., Shen, Q. et al. Gut microbiota in early pregnancy among women with Hyperglycaemia vs. Normal blood glucose. BMC Pregnancy Childbirth 20, 284 (2020). https://doi.org/10.1186/s12884-020-02961-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12884-020-02961-5