
Abstract
Background
We proposed to investigate high-dose pharmaceutical-grade biotin in a population of demyelinating neuropathies of different aetiologies, as a proof-of-concept.
Methods
Phase IIb open label, uncontrolled, single center, pilot study in 15 patients (three groups of five patients) with chronic demyelinating peripheral neuropathy, i.e. chronic inflammatory demyelinating polyradiculoneuropathy, anti-myelin-associated glycoprotein neuropathy and Charcot-Marie-Tooth 1a or 1b. The investigational product was high-dose pharmaceutical-grade biotin (100 mg taken orally three times a day over a maximum of 52 weeks.
The primary endpoint was a 10% relative improvement in 2 of the following 4 electrophysiological variables: motor nerve conduction velocity, distal motor latency, F wave latency, duration of the compound muscle action potential. The secondary endpoints included Overall Neuropathy Limitations Scale (ONLS) score, Medical Research Council (MRC) sum score, Inflammatory Neuropathy Cause and Treatment (INCAT) sensory sum score, 10-m walk test, 6-min walk test, posturography parameters, and nerve excitability variables.
Results
The primary endpoint was reached in one patient. In the full population analysis, some secondary endpoints parameters improved: MRC score, INCAT sensory sum score, 6-min walk distance, strength-duration time constant, and rheobase. There was a positive correlation between the improvement in the 6-min walk distance and the strength-duration time constant. Regarding the safety results, 42 adverse events occurred, of which three were of severe intensity but none was considered as related to the investigational product.
Conclusions
Even if the primary endpoint was not met, administration of high-dose pharmaceutical-grade biotin led to an improvement in various sensory and motor parameters, gait abilities, and nerve excitability parameters. The tolerance of the treatment was satisfactory.
Trial registration
ClinicalTrials.gov Identifier: NCT02967679; date 2016/12/05.
Similar content being viewed by others


Introduction
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), polyneuropathy associated with IgM monoclonal gammopathy with antibodies against myelin-associated glycoprotein (MAG) and Charcot-Marie-Tooth (CMT) 1a and 1b are acquired and genetic neuropathies that alter the myelin sheath as a common feature.
In CIDP, the lesions include myelin sheaths and nodes of Ranvier morphological changes leading to alterations in nerve excitability. These alterations include a decrease in Na + /K + adenosine 5′-triphosphatase (ATPase) pump function, intra-axonal Na + accumulation associated with energetic failure and variable modifications in fast and slow Na + and K + voltage-gated channels [1,2,3,4].
Anti-MAG antibodies lead to enlargement of the myelin sheath [5] The duplication of the PMP22 gene in CMT1a patients, and point mutations in the myelin protein 0 (MPZ) gene in CMT1b patients result in their overexpression and in abnormal Schwann cell and myelin differentiation.
Despite different pathophysiological mechanisms, these neuropathies share common electrophysiological features of demyelination. They also share common clinical features, including variable degrees of progressive weakness, fatigability, or sensory deficit in both upper and lower limbs, ataxia, and impaired walking abilities.
Treatment options for CIDP include steroids, intravenous immunoglobulins and plasma exchange. However, the long-term (more than 6 months) benefits of these therapies may remain limited, leaving room for the development of chronic and severe disability. There is no consensus about the best treatment strategy for anti-MAG patients [6]. There is currently no approved treatment for either CMT1a or for CMT1b.
Biotin (or vitamin H) is a ubiquitous water-soluble vitamin that is naturally found in many foods. In mammals, biotin acts as a coenzyme for four important carboxylases involved in key steps of energy metabolism (Krebs cycle) and fatty acid synthesis. High-dose pharmaceutical-grade biotin (hdPB) has been considered a potential therapeutic option in demyelinating disease of the CNS, i.e., secondary multiple sclerosis (MS) [7, 8], although this was recently refuted [9]. Biotin improved clinical and neurophysiological parameters in a small series of patients [10] and decreased pain in an experimental study [11].
Previous experience in three Phase IIb/III clinical research studies in humans has provided evidence of no risk of hdPB (300 mg/day) in adult MS patients [7,8,9, 12].
We hypothesized that a positive effect of hdPB in demyelinating neuropathies could be associated with (i) an increased energy production in demyelinated nerve fibers that would avoid neurodegeneration and improve neuronal functioning and (ii) a stimulation of myelin repair through activation of the acetyl-CoA carboxylase in Schwann cells.
Considering its previous use in MS, we proposed to study the intake of hdPB in a population of patients with demyelinating neuropathies of different etiologies in a proof of concept/pilot study. To this end, we used clinical scales assessing neurological deficits and function, posturography, and electrophysiological techniques of nerve conduction and excitability studies. We chose a neurophysiological primary endpoint less likely to be influenced by a placebo effect in an open-label, uncontrolled study. Previous studies have shown that, at least in the context of CIDP, neurophysiological criteria have proven to be sensitive to changes induced by therapeutics [13]. Moreover, since the present study focused on the consequences of peripheral nerve demyelination whatever its origin and its clinical results, the objective was to investigate how the treatment could modify the metabolic processes and the dysfunction of nerve fibers whose myelin is impaired.
Patients and methods
Patients
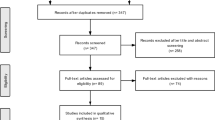
This open-label, uncontrolled, single-center, pilot study (ClinicalTrials.gov Identifier: NCT02967679; date 2016/12/05) included 15 patients in 3 distinct groups of 5 patients suffering from specific etiologies of chronic demyelinating peripheral neuropathy: CIDP (n = 5), anti-MAG (n = 5), CMT1a or CMT1b (n = 5) (Fig. 1). The study protocol, amendments, and patient-related documents, were approved by the Independent Ethics Committee (IEC/IRB) of Ile de France VI – Groupe Hospitalier Pitié-Salpêtrière 47, boulevard de l’hôpital 75,013 Paris, France (n° 128–15, Eudra-CT: 2015–001150-15).. Patients were recruited in the Center of Reference for Neuromuscular Disorders of Henri Mondor University Hospital, Créteil, France. The study period ranged from December 05, 2016, to March 18, 2019. The patients fulfilled the diagnostic criteria of either CIDP [14], anti-MAG polyneuropathy, or genetically defined CMT1a or CMT1b, and gave their written informed consent. Other causes of peripheral neuropathies were excluded on the basis of negative results of a large battery of investigations. Absence of neurotoxic drug intake was also checked. Disease-modifying treatment (intravenous immunoglobulins for the 5 patients with CIDP) and symptomatic treatments were not modified during a six-month period preceding the study and during the study period. No medical or surgical history could interfere with the study investigations. No previous or concomitant treatment was considered to interfere with the results of the study.

Participant flowchart
The main non inclusion criteria consisted of pregnancy, women of childbearing potential without effective contraception, and relapse in the past 3 months before inclusion for CIDP patients.
Treatment
Capsules of hdPB consisting of 100 mg biotin and excipients (lactose, magnesium stearate, croscarmellose sodium, silica) (MD1003, Medday Pharmaceutical) were taken orally three times a day (one in the morning, one at noon, and one in the evening) over a maximum of 52 weeks.
Study schedule and procedures
The study schedule consisted of screening and inclusion visits, which were merged, followed by 4 visits at three months intervals over 52 weeks, whereas the initial schedule planned for visits every 12 weeks over 48 weeks. Clinical and functional data were obtained every 12 weeks (baseline W0, W12, W24, W36, W48). Neurophysiological examination, postural stability, and biological testing were performed every 24 weeks (W0, W24, W48).
Clinical information
The following information was recorded: (i) clinical data (demographic data, history of the patient’s disease, other relevant medical history and ongoing concomitant diseases, clinical examination, vital signs: systolic and diastolic blood pressure, temperature, heart rate, concomitant therapies); (ii) clinical scores (Medical Research Council (MRC) sum score including 19 muscles evaluated bilaterally (max: 190), Inflammatory Neuropathy Cause and Treatment (INCAT) sensory sum score) and functional assessment (timed 10-m walk test, 6-min walk test, Overall Neuropathy Limitations Scale (ONLS) score) [15,16,17].
Neurophysiological examination
A conventional motor nerve conduction study was performed as follows: two motor nerves were explored at each upper limb (median and ulnar nerves bilaterally) and each lower limb (peroneal and tibial nerves bilaterally); compound muscle action potentials were recorded at both hands and feet, and also at the leg (tibialis anterior muscle) for the peroneal nerve; two or three sensory nerves were explored at each upper limb (median, ulnar, and radial nerves bilaterally) and each lower limb (peroneal and sural nerves bilaterally). The recorded parameters were: amplitude (peak-to-peak) and duration (negative peak) of the compound muscle action potentials, distal motor latency, motor nerve conduction velocity, and F wave latency for motor nerves, amplitude of sensory nerve action potentials and sensory nerve conduction velocity for sensory nerves. A minimal amplitude was set at 100 µV for compound muscle action potentials and 1 µV for sensory nerve action potentials. Below these values, the potentials were considered as absent (0 µV). The same protocol was applied at each visit for each patient.
In the same session, a protocol of nerve excitability testing was also performed, including the measurement of the following variables: minimal and maximal absolute refractory period duration (ms), refractoriness (%), supernormality (%), strength-duration time constant (SDTC) (ms), and rheobase (mA).
The minimal and maximal absolute refractory period durations were measured according to the double-collision method [18]. The percentages of refractoriness and supernormality were calculated at interstimulus intervals of 2 ms and 7 ms, respectively [19]. Strength–duration properties were determined by measuring the threshold intensities required to obtain 70% Mmax at 0.1-ms and 1.0-ms pulse durations. The strength–duration time constant (chronaxie) and rheobase were calculated using formulae derived from Weiss’s Law [20].
Spatial–temporal walking analysis
All patients were asked to walk 10 m, barefoot without assistance, on a pressure sensitive carpet (GaitRite Platinum, size 0.61*7.92 m, sample frequency 60 Hz, CIR Systems, Inc., Sparta, NJ, USA), twice at their comfortable and twice at maximal walking velocity. Eight key parameters were calculated over the 7.92 m in the middle of the walkway (excluding the acceleration and deceleration phases) in each condition: mean of speed (m/s), cadence (step/s), step length (m), coefficient of variation of step length, spatial asymmetry index [21], duration of foot support in percentage of the walking cycle (%), step width (m), and foot angle versus walking axis (°).
Postural stability
Participants stood upright barefoot on a force plate (size 400*600 mm, sample frequency 1000 Hz, Bertec Corporation, Columbus, OH, USA) during 60 s in four conditions: eyes open and eyes closed, in 2 standardized positions: feet apart (distance between the calcaneus, 16 cm; angle of the foot, 17°) [22] and feet together. The following dependent variables were extracted from the center of pressure (COP) trajectories: the range of COP positions (i.e. the difference between maximum and minimum COP positions) computed in both anteroposterior (AP range, mm) and medio-lateral directions (ML range, mm); the ellipse best fitting the planar COP data and covering 95% of COP positions (Area, mm2) calculated based on a principal component analysis that determined the alignment of its major and minor axes; the mean planar velocity (Velocity, mm/s) calculated as the sum of Euclidian distances between successive positions (total length) divided by the sample duration [23].
Biological sampling
Biological investigation included a battery of safety and etiological parameters at the time of inclusion (see Online Only Supplements). For women of childbearing potential: a highly sensitive pregnancy test (human chorionic gonadotropin HCG) was performed.
Evaluation criteria
Primary endpoint
The primary endpoint was determined on four motor nerve conduction study variables: motor nerve conduction velocity, distal motor latency, F wave latency, and compound muscle action potential duration. The percent of improvement from baseline to the end of the study (W48) was calculated for each neurophysiological variables and for each of the 8 explored motor nerves (median, ulnar, peroneal, and tibial nerves bilaterally). An improvement ≥ 10% for at least 3 of the 8 explored nerves was arbitrary considered relevant. The primary endpoint was met in a given patient if such a 10% improvement was achieved in at least 2 of the 4 motor nerve conduction study variables.. We assumed that such a change could be considered as really significant and meaningful.
Secondary endpoints
Changes from W0 to W48 in the following clinical and electrophysiological parameters:
Clinical endpoints: ONLS score, MRC sum score and total score, INCAT Sensory Sum Score, timed 10-m walk test (s), 6-min walk test distance (m), postural stability parameters and spatial–temporal walking parameters.
Nerve excitability endpoints: minimal and maximal absolute refractory period duration (ms), refractoriness (%), supernormality (%), SDTC (ms), and rheobase (mA).
Safety criteria
Analysis of extent of exposure, treatment-emergent adverse events (TEAEs) and laboratory testing.
Statistical analysis
The study included 15 patients in total divided into 3 subgroups of 5 patients suffering from either CIDP, anti-MAG or CMT1a/CMT1b. It was arbitrary assumed that, in a pilot proof-of-concept study, 15 patients were a sufficient sample to detect clinical or electrophysiological changes related to hdPB intake or to show the best endpoints for possible future confirmatory studies in the context of the most common types of chronic demyelinating peripheral neuropathies.
The primary analysis of the primary endpoint was evaluated on the full analysis set (FAS) population. Missing values at W48 for the four criteria of demyelination were imputed using the last observation carried forward (LOCF) method. The same analysis as the sensitivity analysis was performed on the per-protocol set population. As a secondary analysis of the primary endpoint, a Wilcoxon signed rank test was performed on the four criteria of demyelination to assess the significance of the relative change from W0 to W48 on the FAS, considering all nerves pooled per patient and criterion. Each test was performed at a 2-sided significance level of 5%. A Wilcoxon signed rank test was performed overall for each parameter to assess whether the absolute changes from W0 were significant at W48. Secondary endpoints were evaluated based on the absolute change from W0 to W48 by disease groups on the FAS. Missing values at W48 were imputed using the LOCF method. A Wilcoxon signed rank test was performed overall for each parameter to assess whether the absolute changes from W0 were significant at W48. We used a Pearson test, after checking the normal distribution of values by the Kolmogorov–Smirnov test, to study the correlation concerning the significant changes observed from W0 to W48 between clinical and neurophysiological data.
Results
Patients
All 15 patients allocated to treatment were included in the FAS and safety populations. Seven participants (47%) presented major deviations and were excluded from the per-protocol set population: 3 in the CIDP group, 3 in the anti-MAG group and 1 in the CMT1 group. Major deviations were as follows: compliance not between 80 and 120% (1 patient with anti-MAG); patients without electrophysiological parameters worsening for the past 3 years (7 patients: CIDP, n = 3, anti-MAG, n = 3 and CMT1, n = 1); missing evaluation at W0 or at W48 for motor nerve conduction parameters (1 patient with CIDP); hdPB (investigation medical product) administration duration different from 336 ± 15 days (1 patient with anti-MAG); hdPB administration interruption > 10 consecutive days (1 patient with anti-MAG on his own decision; see supplementary material eTable 1). Demographic and clinical data are presented in Table 1.
Efficacy results
The primary endpoint (primary analysis) was reached in the FAS in 1 (CMT1) patient. This was confirmed in the per-protocol set population. More precisely, in the FAS population, improvements of at least 10% in at least 3 out of 8 nerves were observed in motor nerve conduction velocity in 2 patients, distal motor latency in 4 patients, F wave latency in 0 patients, and compound muscle action potential duration in 4 patients (Tables 2 and 3).
In the overall FAS population, some noteworthy secondary endpoint parameters showed improvements, as described below. All results are expressed as median interquartile ranges (IQR) (1st quartile;3rd quartile).
Clinical parameters
The MRC total score increased from 168.0 (160.0;177.0) at W0 to 176.0 (166.0;180.0) at W48 (absolute change, 3.0 (0.0;9.0), p = 0.0371), the INCAT sensory sum score decreased from 6.0 (4.0;8.0) at W0 to 3.0 (1.0;7.0) at W48 (absolute change, -3.0 (-3.0;0.0), p = 0.0142), and the 6-min walking distance increased from 375.0 (293.0;474.0) m at W0 to 469.0 (278.0;506.0) m at W48 (absolute change, 69.0 (10.0;104.0) m, p < 0.001; Tables 4 and 5).
Spatial–temporal walking analysis
Improvement of the walking ability between W0 and W48 was observed regarding cadence from 1.76 (1.66;1.82) step/s at W0 to 1.91 (1.78;1.98) step/s at W48 (absolute change, 0.08 (0.00;0.16) step/s, p = 0.0225), duration of right foot support from 0.57 (0.56;0.60) s at W0 to 0.52 (0.50;0.57) s at W48 (absolute change, -0.03 (0.00;0.16) s, p = 0.0137) and duration of left foot support from 0.57 (0.54;0.60) s at W0 to 0.53 (0.50;0.56) s at W48 (absolute change, -0.02 (-0.08;0.00) s, p = 0.0322) in ‘comfortable speed’ condition.
Postural stability
The posturography parameters remained unchanged between W0 and W48.
Excitability parameters
Median (IQR) SDTC values increased from 0.13 (0.12;0.36) ms at W0 to 0.32 (0.14;0.36) ms at W48 (absolute change of 0.05 (0.01;0.14) ms, p = 0.039); rheobase values decreased from 12.50 (7.02;20.61) mA at W0 to 7.31 (3.72;11.39) mA at W48 (absolute change of -4.21 (-9.22;-0.78) mA, p < 0.001) (Tables 6, 7 and 8).
Clinical and neurophysiological correlations
We studied the correlations between the parameters that significantly changed from W0 to W48, i.e. the 6-min walk test for clinical parameters and SDTC and rheobase for electrophysiological parameters. There was a positive correlation between SDTC and walking capacities evaluated by the 6-min walk test (r = 0.579; p = 0.049) but no correlation between rheobase and the 6-min walk test (r = -0.203; p = 0.528).
Safety results
Overall, 14 patients presented 42 TEAEs (eTable 2). Three TEAEs were of severe intensity in 3 patients, including 1 patient in each disease group: a relapsing autoimmune encephalopathy occurred 428 days after hdPB discontinuation in a patient with CIDP, leading to death; one renal clear cell carcinoma occurred in a patient with anti-MAG neuropathy; and left external malleolar fracture occurred in a patient with CMT1. None of these events was considered to be related to hdPB treatment.
Laboratory test interferences due to hdPB were reported in 2 patients, leading to incorrect interpretation of thyroid tests.
All other TEAEs were mild to moderate in intensity (see eTable 2) without relevant differences between disease groups. No clinically relevant shifts in biological parameters were observed during the study.
Discussion
This phase IIb open-label trial investigating the effect of hdPB in demyelinating peripheral neuropathies did not reach the primary endpoint, which consisted of a composite nerve conduction study criterion. However, several clinical, functional, and neurophysiological secondary endpoints were achieved. Improvements in clinical deficits and disability were observed, as well as changes in nerve excitability parameters, i.e., SDTC (chronaxie) increase and rheobase decrease. Moreover, we found a positive correlation between improvements in the 6-min walk test and the SDTC.
Standard nerve conduction studies did not show any modification in the overall population, whereas excitability parameter changes occurred, regardless the small sample size. These two types of electrophysiological studies do not assess the same aspects of nerve function and certainly do not have the same sensitivity to changes induced by therapy. Nerve conduction study parameters is a surrogate of the propagation of nerve impulses and the structural properties of axons, while nerve excitability parameters are related to physiological properties of axonal membrane potential changes, involving various slow and fast KV channels, slow NaV channels and the Na + /K + ATPase pump [3, 24,25,26]. Different alterations of these nerve excitability parameters have been reported in demyelinating neuropathies of acquired or genetic origin [3, 27]. Excitability studies are an interesting way to assess drug-induced changes in nerve function. For example, excitability changes have been observed shortly after IVIg and following intravenous immunoglobulin infusions [4, 13].
In this study, the main changes in nerve excitability after hdPB intake were a decrease in rheobase and an increase in chronaxie (SDTC), two variables whose reproducibility has been shown in several studies, including those performed in our laboratory [28,29,30]. In CIDP, rheobase is known to be increased and SDTC to be decreased [31], while no such changes have been previously reported in patients with anti-MAG neuropathy and CMT1 [32, 33]. Chronaxie and rheobase mainly reflect nodal aspects of resting membrane potential and depend on the amount of persistent Na + currents through the slow NaV channels. A reduced rheobase and increased SDTC, as observed after hdPB treatment, suggest an increased expression or activity of slow NaV channels [34, 35]. An inward persistent Na + flow results in depolarizing changes in resting membrane potential [36,37,38]. This means that axonal excitability has increased after hdPB treatment and the current influx required to elicit action potentials and ensure nerve conduction was lower. The expression of NaV channels (NaV 1.2 and NaV 1.6) at the origin of persistent Na + currents was shown to be modified in demyelinated axons in the context of MS [38]. Therefore, our results suggest that a treatment with hdPB could generate a phenomenon of nerve plasticity at the axonal level, a potential therapeutic target of nerve excitability in demyelinating neuropathies.
Motor and sensory deficits, as assessed by the MRC total score and the INCAT sensory sum score, improved after treatment. The median distance in the 6-min walking test increased by 69 m, and other clinical and posturography parameters improved. The walking capacity increased by more than 0.5 standard deviations, which is considered a clinically meaningful difference [39]. Although improvement in walking capacities in an open-label, uncontrolled study may be related to important bias, the positive correlation with changes in a nerve excitability parameter (SDTC) that is related to the intrinsic properties of the nerve fibers reinforces the hypothesis of a real effect of the drug. In the context of MS treatment with hdPB, whose initially reported beneficial effects have not been confirmed [9], we did not observe changes in synaptic excitability in the evaluation of the central neural networks [41]. This obviously does not exclude the possibility of excitability changes following hdPB intake at the level of peripheral axons.
Overall, the treatment with hdPB was well tolerated in the 15 patients included in this study. An 80-year-old patient developed an inflammatory encephalopathy with a relapse 428 days after treatment interruption. Although the occurrence of an autoimmune process is surprising at this age [40,41,42], the relapse that occurred more than one year after drug discontinuation precludes any direct relationship with the treatment. Moreover, in a previous study of hdPB in MS, no central nervous system inflammatory event was observed [9, 12, 43]. Thus, in this study, no new adverse events of hdPB treatment were observed, apart from those already known and reported [7].
The present study has several limitations. First, this is not a placebo-controlled trial, and the positive clinical results may be in part related to a placebo effect, even if they are reinforced by the neurophysiological changes. The number of patients included in the study was small, and a larger study will be required to validate the present observations. Moreover, the population was not homogeneous, with different mechanisms of myelin alterations between the patient groups.
Conclusions
The present results, especially nerve excitability changes and clinically meaningful improvement in currently less treatable or untreatable conditions represented by anti-MAG neuropathy and CMT1a support the construction of a large randomized controlled trial to assess the potential benefit of treating demyelinating neuropathies with hdPB.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ATPase:
-
Adenosine 5′-triphosphatase
- CIDP:
-
Chronic inflammatory demyelinating polyradiculoneuropathy
- CMT:
-
Charcot-Marie-Tooth
- COP:
-
Center of pressure
- FAS:
-
Full analysis set
- HdPB:
-
High-dose pharmaceutical-grade biotin
- IEC/IRB:
-
Independent Ethics Committee
- INCAT:
-
Inflammatory Neuropathy Cause and Treatment
- LOCF:
-
Last observation carried forward
- MAG:
-
Myelin-associated glycoprotein
- MPZ:
-
Myelin protein 0
- MRC:
-
Medical Research Council
- MS:
-
Multiple sclerosis
- ONLS:
-
Overall Neuropathy Limitations Scale (ONLS)
- SDTC:
-
Strength-duration time constant
- TEAEs:
-
Treatment-emergent adverse events
References
Stys PK, Waxman SG, Ransom BR. Na(+)-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol. 1991;30(3):375–80.
Smith KJ, Kapoor R, Hall SM, Davies M. Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol. 2001;49(4):470–6.
Lefaucheur JP, Boërio D, Hogrel JY, Créange A. Nerve excitability studies in the assessment of dysimmune neuropathies. Rev Neurol (Paris). 2006;162(Suppl. 1). Spec No 1:3S17–3S26.
Boerio D, Creange A, Hogrel JY, Gueguen A, Bertrand D, Lefaucheur JP. Nerve excitability changes after intravenous immunoglobulin infusions in multifocal motor neuropathy and chronic inflammatory demyelinating neuropathy. J Neurol Sci. 2010;292(1–2):63–71.
European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society--First Revision. J Peripher Nerv Syst 2010;15(1):1–9.
Lunn MP, Nobile-Orazio E. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;5:CD002827.
Sedel F, Papeix C, Bellanger A, Touitou V, Lebrun-Frenay C, Galanaud D, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159–69.
Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2016;110(Pt B):644–53.
Cree BAC, Cutter G, Wolinsky JS, Freedman MS, Comi G, Giovannoni G, et al. Safety and efficacy of MD1003 (high-dose biotin) in patients with progressive multiple sclerosis (SPI2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19:988–97.
Koutsikos D, Agroyannis B, Tzanatos-Exarchou H. Biotin for diabetic peripheral neuropathy. Biomed Pharmacother. 1990;44(10):511–4.
Abed AR, Abed A, Banafshe HR, Malekabad ES, Gorgani-Firuzjaee S, Dadashi AR. Effect of biotin supplementation on neuropathic pain induced by chronic constriction of the sciatic nerve in the rat. Res Pharm Sci. 2021;16(3):250–9.
Mathais S, Moisset X, Pereira B, Taithe F, Ciron J, Labauge P, et al. Relapses in Patients Treated with High-Dose Biotin for Progressive Multiple Sclerosis. Neurotherapeutics. 2021;18(1):378–86.
Lin CS-Y, Krishnan AV, Park SB, Kiernan MC. Modulatory Effects on Axonal Function After Intravenous Immunoglobulin Therapy in Chronic Inflammatory Demyelinating Polyneuropathy. Arch Neurol. 2011;68(7):862–9.
Joint Task Force of the EFNS and the PNS. J Peripher Nerv Syst. 2010;15(1):1–9.
Graham RC, Hughes RA. A modified peripheral neuropathy scale: the Overall Neuropathy Limitations Scale. J Neurol Neurosurg Psychiatry. 2006;77(8):973–6.
Merkies IS, Schmitz PI, Van der Meche FG, Van Doorn PA. Psychometric evaluation of a new sensory scale in immune-mediated polyneuropathies. Inflammatory Neuropathy Cause and Treatment (INCAT) Group. Neurology. 2000;54(4):943–9.
Medical research council. Aids to the investigation of peripheral nerve injuries. 2nd ed.; 1943.
Ingram DA, Davis GR, Swash M. The double collision technique: a new method for measurement of the motor nerve refractory period distribution in man. Electroencephalogr Clin Neurophysiol. 1987;66(3):225–34.
Kiernan MC, Mogyoros I, Burke D. Differences in the recovery of excitability in sensory and motor axons of human median nerve. Brain. 1996;119:1099–105.
Bostock H, Bergmans J. Post-tetanic excitability changes and ectopic discharges in a human motor axon. Brain. 1994;117:913–28.
Plotnik M, Giladi N, Hausdorff JM. A new measure for quantifying the bilateral coordination of human gait: effects of aging and Parkinson’s disease. Exp Brain Res. 2007;181(4):561–70.
McIlroy WE, Maki BE. Preferred placement of the feet during quiet stance: development of a standardized foot placement for balance testing. Clin Biomech (Bristol, Avon). 1997;12(1):66–70.
Albertsen IM, Ghédira M, Gracies JM, Hutin É. Postural stability in young healthy subjects - Impact of reduced base of support, visual deprivation, dual tasking. J Electromyogr Kinesiol. 2017;33:27–33.
Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve. 1998;21(2):137–58.
Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve. 2000;23(3):399–409.
Krishnan AV, Lin CS, Park SB, Kiernan MC. Assessment of nerve excitability in toxic and metabolic neuropathies. J Peripher Nerv Syst. 2008;13(1):7–26.
Krishnan AV, Lin CS, Kiernan MC. Activity-dependent excitability changes suggest Na+/K+ pump dysfunction in diabetic neuropathy. Brain. 2008;131(Pt 5):1209–16.
Boerio D, Hogrel JY, Creange A, Lefaucheur JP. A reappraisal of various methods for measuring motor nerve refractory period in humans. Clin Neurophysiol. 2005;116(4):969–76.
Mogyoros I, Lin C, Dowla S, Grosskreutz J, Burke D. Reproducibility of indices of axonal excitability in human subjects. Clin Neurophysiol. 2000;111(1):23–8.
Boerio D, Hogrel JY, Lefaucheur JP, Wang FC, Verschueren A, Pouget J, et al. Stimulus-response curve of human motor nerves: multicenter assessment of various indexes. Neurophysiol Clin. 2008;38(1):31–8.
Cappelen-Smith C, Kuwabara S, Lin CS, Mogyoros I, Burke D. Membrane properties in chronic inflammatory demyelinating polyneuropathy. Brain. 2001;124(Pt 12):2439–47.
Nodera H, Bostock H, Kuwabara S, Sakamoto T, Asanuma K, Jia-Ying S, et al. Nerve excitability properties in Charcot-Marie-Tooth disease type 1A. Brain. 2004;127(Pt 1):203–11.
Garg N, Park SB, Howells J, Noto YI, Vucic S, Yiannikas C, et al. Anti-MAG neuropathy: Role of IgM antibodies, the paranodal junction and juxtaparanodal potassium channels. Clin Neurophysiol. 2018;129(10):2162–9.
Mogyoros I, Kiernan MC, Burke D. Strength-duration properties of human peripheral nerve. Brain. 1996;119(Pt 2):439–47.
Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol. 1997;498(Pt 1):277–94.
Krishnan AV, Lin CS, Park SB, Kiernan MC. Axonal ion channels from bench to bedside: a translational neuroscience perspective. Prog Neurobiol. 2009;89(3):288–313.
Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol. 2001;112(9):1575–85.
Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101(21):8168–73.
Revicki D, Hays RD, Cella D, Sloan J. Recommended methods for determining responsiveness and minimally important differences for patient-reported outcomes. J Clin Epidemiol. 2008;61(2):102–9.
Algahtani H, Shirah B, Alassiri A. Tumefactive demyelinating lesions: A comprehensive review. Mult Scler Relat Disord. 2017;14:72–9.
Brod SA, Lindsey JW, Nelson F. Tumefactive demyelination: Clinical outcomes, lesion evolution and treatments. Mult Scler J Exp Transl Clin. 2019;5(2):2055217319855755.
Ohe Y, Hayashi T, Mishima K, Nishikawa R, Sasaki A, Matsuda H, et al. Central nervous system lymphoma initially diagnosed as tumefactive multiple sclerosis after brain biopsy. Intern Med. 2013;52(4):483–8.
Kauv P, Chalah M, Créange A, Ayache S, Hodel J. No change in phosphorous magnetic resonance spectroscopy and diffusion tensor imaging in patients with progressive multiple sclerosis treated with high-dose biotin. Berlin: A one-year follow-up study. ECTRIMS; 2018.
Acknowledgements
Not applicable.
Funding
Medday Pharmaceuticals.
Author information
Authors and Affiliations
Contributions
AC performed literature search, clinical analysis and management, collected, analyzed and interpreted data, advised on design or analysis and wrote paper, EH performed literature search, clinical analysis and management, collected, analyzed and interpreted data, advised on design or analysis and wrote paper, FS performed literature search advised on design or analysis, LLV wrote the statistical plan analyzed and interpreted data, JPL performed literature search, clinical analysis and management, collected, analyzed and interpreted data, advised on design or analysis and wrote paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol, amendments, and patient-related documents, were approved by the Independent Ethics Committee (IEC/IRB) of Ile de France VI – Groupe Hospitalier Pitié-Salpêtrière 47, boulevard de l’hôpital 75013 Paris, France (n° 128–15, Eudra-CT: 2015–001150-15).
Written informed consent was obtained from all the participants.
All experiments were performed in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
AC reports grants and nonfinancial support from Medday, during the conduct of the study; personal fees from Medday, personal fees from Merck, grants from Octapharma, grants and personal fees from Novartis, grants and personal fees from Roche, and grants and personal fees from Biogen, outside the submitted work.
EH has nothing to disclose.
FS was employee and shareholder of Medday; personal fees from medday pharmaceuticals, outside the submitted work; In addition, FS has a patent Pharmaceutical Compositions highly dosed with Biotin EP2555771/ US 20130084334 with royalties paid to Medday, a patent Method of treating Amyotrophic Lateral Sclerosis and neuropathies with Biotin EP 3273957/ US 20190314342 issued, and a patent Methods of Treating Multiple Sclerosis with Biotin EP2729143/US 20140030331 with royalties paid to Medday.
LLV is an employee of ATLANSTAT.
JPL has nothing to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
eTable 1. Compliance to IP During the Whole Study - SS population. eTable 2. Treatment-emergent Adverse Events (TEAEs). eTable 2a. Overview of the incidence of TEAEs - SS Population. eTable 2b. Display of Adverse Events. eTable 2c. IP-related TEAEs by SOC and PT – SS Population. eTable 2d. TEAEs by IP Discontinuation by SOC and PT – SS Population. eTable 2e. TEAEs Leading to Death by SOC and PT – SS Population. eTable 2f. Other Serious Adverse Events by SOC and PT – SS Population.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Créange, A., Hutin, E., Sedel, F. et al. High-dose pharmaceutical-grade biotin in patients with demyelinating neuropathies: a phase 2b open label, uncontrolled, pilot study. BMC Neurol 23, 389 (2023). https://doi.org/10.1186/s12883-023-03440-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-023-03440-y