
Abstract
Background
Spastic paraplegia type 4 (SPG4) is caused by mutations in the SPAST gene, is the most common form of autosomal-dominant pure hereditary spastic paraplegias (HSP), and is rarely associated with a complicated form that includes ataxia, epilepsy, and cognitive decline. To date, the genotype-phenotype correlation has not been substantially established for SPAST mutations.
Case presentation
We present a Japanese patient with infantile-onset HSP and a complex form with coexisting ataxia and epilepsy. The sequencing of SPAST revealed a de novo c.1496G > A (p.R499H) mutation. A review of the literature revealed 16 additional patients with p.R499H mutations in SPAST associated with an early-onset complicated form of HSP. We found that the complicated phenotype of patients with p.Arg499His mutations could be mainly divided into three subgroups: (1) infantile-onset ascending hereditary spastic paralysis, (2) HSP with severe dystonia, and (3) HSP with cognitive impairment. Moreover, the c.1496G > A mutation in SPAST may occur as a de novo variant at noticeably high rates.
Conclusion
We reviewed the clinical features of the patients reported in the literature with the p.Arg499His mutation in SPAST and described the case of a Japanese patient with this mutation presenting a new complicated form. Accumulating evidence suggests a possible association between infantile-onset complicated HSP and the p.Arg499His mutation in SPAST. The findings of this study may expand the clinical spectrum of the p.Arg499His mutation in SPAST and provide an opportunity to further study the genotype-phenotype correlation of SPG4.
Similar content being viewed by others


Background
Hereditary spastic paraplegias (HSPs) are clinically and genetically heterogeneous neurodegenerative disorders characterized by progressive weakness and spasticity in the lower limbs due to pyramidal tract dysfunction [1]. HSPs can be inherited in an autosomal-dominant (AD), autosomal-recessive (AR), X-linked, or mitochondrial manner [2]. An isolated pyramidal syndrome affecting predominantly in the lower limbs with or without vibration sense impairment and urinary urgency defines pure HSP, whereas complicated HSP presents a more complex clinical picture with additional neurological findings, including ataxia, epilepsy, and cognitive decline [1, 2]. AR-HSP is usually a complicated form, contrary to AD-HSP, which is mostly a pure form [3].
Spastic paraplegia type 4 (SPG4) is due to heterozygous mutations in the SPAST gene and is the most frequent cause of both familial and sporadic HSP [1]. SPG4 in most cases is considered a pure HSP. SPG4 is rarely associated with additional neurological signs [4]. Here, we present the case of a Japanese patient with a complex form of infantile-onset HSP with coexisting ataxia and epilepsy. SPAST sequencing revealed a de novo c.1496G > A (p.R499H) mutation. A review of the literature revealed 16 additional patients with p.R499H mutations in SPAST associated with early-onset complicated forms of HSP.
Case presentation
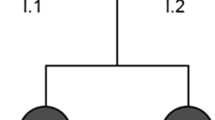
A 4-year-old girl (Fig. 1A, II-3) was the third of three siblings born to healthy and unrelated parents. Her 8-year-old and 5-year-old sisters (Fig. 1A II-1 and II-2) were unaffected. She was born by vaginal delivery after an uneventful pregnancy. Her parents initially became concerned when she could not crawl on her hands and knees or stand unassisted by 17 months of age. She could lift her head by 4 months, roll over by 6 months, sit up unsupported by 10 months, crawl on her belly by 11 months, stand with assistance by 18 months, and walk with assistance by 22 months. She could not walk independently until now. A neurological examination at 17 months of age revealed slight muscle weakness in the distal lower extremities and exaggerated deep tendon reflexes in the lower limbs. She started rehabilitation therapy at that point. At 2 years and 3 months, she presented a scissor gait when walking with assistance. At 2 years and 5 months, truncal ataxia was detected. She was intellectually normal, and no nystagmus, intention tremor, dysmetria, or speech impairment was detected. At 3 years and 3 months, the Kyoto Scale of Psychological Development (KSPD) test revealed no cognitive developmental delay, with the following scores: Postural-Motor functions, 26; Cognitive-Adaptive functions, 96; and Language-Social functions, 81. A nerve conduction study revealed nothing abnormal. Metabolic and routine blood investigations were unremarkable.

A Pedigree of the present family. The proband is indicated (arrow). Squares indicate men, circles women, shaded (black) symbols individuals with symptoms of complicated HSP, and unshaded ones individuals without any symptoms. Individuals evaluated both clinically and genetically are denoted by asterisks. B Sanger sequencing revealed the c.1496G > A mutation in SPAST was in a heterozygous state in the proband. The c.1496 nucleotide is shaded yellow. C The c.1496G > A mutation in SPAST was not detected in the proband’s father. The c.1496 nucleotide is shaded yellow. D The c.1496G > A mutation in SPAST was not detected in the proband’s mother. The c.1496 nucleotide is shaded yellow. E The c.1496G > A mutation in SPAST was not detected in the proband’s younger sister. The c.1496 nucleotide is shaded yellow
At 3 years and 5 months, she had an unprovoked epileptic seizure with forced rightward eye deviation. Although acute symptomatic seizures did not occur after that seizure, nonsymptomatic electroencephalogram (EEG) abnormalities with centroparietal spikes were repeatedly recorded. Epilepsy was diagnosed, and she was started on levetiracetam, which she continues to take. The abnormal EEG wave disappeared after antiepileptic treatment, and has been seizure-free ever since.
The motor symptoms of the patient progressed slowly, and her gait became increasingly slow and spastic over time. On neurological examination at age 4, she presented slight paresis, increased muscle reflexes, and increased muscle tone and stiffness in all four limbs. Knee and ankle clonus and bilateral Babinski signs were detected. Truncal ataxia, intention tremor, and a staggering gait were also detected. There were no speech or eye movement abnormalities. Magnetic resonance imaging (MRI) of the brain and spine performed at ages 2, 3, and 4 revealed no abnormalities.
We carried out whole-exome sequencing of genomic DNA from the patient. Genomic DNA was extracted from peripheral blood. Exome capture was performed with a SureSelect Human All Exon V6 + UTR (89 Mb) Kit (Agilent Technologies, Santa Clara, CA, USA). Paired-end sequencing was carried out on a HiSeq2500 (Illumina, San Diego, CA, USA) using a HiSeq SBS Kit V4 (Illumina), which generated 100-bp reads. The reference databases utilized included hg38 (GRCh38) (http://genome.ucsc.edu), HGMD (https://portal.biobase-international.com), gnomAD (http://gnomad.broadinstitute.org), and dbSNP (https://www.ncbi.nlm.nih.gov/SNP). We examined variants of 250 genes known to be responsible for or associated with spinocerebellar ataxia, Charcot-Marie-Tooth disease, or HSP (Additional file 1). Through this analysis, we identified a heterozygous missense mutation (c.1496G > A, p.Arg499His) in exon 13 of the SPAST gene in the patient and ruled out the possibility of other causative genes. We then examined exon 13 of the SPAST gene in the patient as well as the patient’s father (Fig. 1A, I-1), mother (Fig. 1A, I-2), and younger sister (Fig. 1A, II-2) by Sanger sequencing. On Sanger sequencing, we reconfirmed the p.R499H mutation in exon 13 of the SPAST gene, which was in a heterozygous state in the patient (Fig. 1B). On the other hand, the patient’s parents and younger sister did not have the mutation (Fig. 1C, D, E). In this family, the patient harbored a mutation that was absent in her parents and her healthy sibling. Her oldest sister was also healthy. These observations suggest that the mutation occurred de novo in the patient.
Discussion and conclusion
The p.R499H mutation in the SPAST gene has been reported as a disease-causing mutation in many previous studies [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. This mutation is located in the AAA ATPase cassette of spastin (from amino acids 342 to 616), which is crucial for microtubule-severing activity [4]. The clinical features of patients with the p.Arg499His mutation reported in the literature are briefly summarized in Table 1.
Including our patient, clinical information is available for 24 patients from different parts of the world. Among them, seventeen patients presented a complicated phenotype, while only 7 were reported to have a pure type of HSP. Admittedly, SPG4 patients with complicated phenotypes are more likely to be reported by clinicians, and our review is strictly not epidemiological. However, it is noteworthy that patients with p.Arg499His mutations are significantly more frequently associated with complicated phenotypes than other mutations in SPAST. Additionally, the patients with this mutation almost invariably also suffered from a more severe type of spastic paraplegia with onset in the first or second year of life. The complicated phenotypes of patients with p.Arg499His mutations can be primarily divided into three subgroups: (1) infantile-onset ascending hereditary spastic paralysis (Patients 1, 6, 7, 9, 10, 14, and 15). These patients all presented very-early-onset (< 2 years old) spastic paraplegia with an ascending phenotype with bulbar involvement mostly occurring during their first decades. (2) Patients 8, 16, 17, and 20 presented early-onset spastic paraplegia with dystonia. (3) Patients 3, 4, 13, and 23 presented early-onset spastic paraplegia with cognitive impairment. Apart from our patient, only one patient (Patient 12) was reported to have associated trunk ataxia in the literature [10]. No further information was provided for that patient; however, both patients presented trunk ataxia as the key symptom of cerebellar dysfunction. Three of the 23 patients (Patients 6, 7, and 14) reported in the literature also had seizures, while Patients 6 and 7 also had an intellectual disability. In contrast, in our patient, cognitive ability was preserved, as evaluated by the KSPD test. Interestingly, most of the patients (19 out of 24) had a de novo mutation, and only 4 were reported to have a family history. This is partly because most patients with the p.Arg499His mutation in SPAST presented a very severe phenotype with a very early onset age. Moreover, this mutation is located in the AAA domain of the SPAST gene, where mutations are clustered [10]. Nevertheless, it is worth noting that the c.1496G > A mutation in SPAST may occur as a de novo variant at noticeably high rates.
We have no explanation for the fact that the p.Arg499His mutation in SPAST can lead to pure HSP as well as to the highly complex phenotypes presented here. The most recognized genetic modifier, c.131C > T (p.Ser44Leu), in SPAST was negative in our patient [4]. There might be some unknown genetic etiology or environmental modifiers underlying the complicated symptoms that could not be identified with the present methods. To date, the genotype-phenotype correlation has not been substantially established for SPAST mutations [4]. However, accumulating evidence suggests that the p.Arg499His mutation in SPAST is associated with severe infantile-onset complicated HSP. Whether the etiology of SPG4 is haploinsufficiency or toxic gain-of-function properties of the mutant spastin proteins remains controversial. The evidence gathered here is not indicative of haploinsufficiency as the cause of the disease since certain mutations in SPAST are more dangerous than others, which would support a toxic gain-of-function etiology, at least in certain missense mutations. Future studies will hopefully allow the further delineation of the complex phenotype associated with the p.Arg499His mutation and the understanding of the underlying molecular mechanisms.
In conclusion, we recognized a complex SPG4 phenotype with infantile-onset severe spastic paraplegia complicated by cerebellar ataxia and epilepsy. This specific phenotype has not previously been reported in the clinical spectrum of SPG4 patients with p.Arg499His mutations. Currently, our patient may still be too young for final phenotyping and thus will need to be followed carefully. A review of the literature revealed Arg499His may occur as a de novo variant at noticeably high rates. The patient we reported here, together with the cases previously reported in the literature, suggests an association between infantile-onset complicated HSP and the p.Arg499His mutation in SPAST. This study may provide an opportunity to further study the genotype-phenotype correlation of SPG4 and shed light on clinicians performing genetic testing for patients with early-onset complicated HSP.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- HSP:
-
Hereditary spastic paraplegias
- AD:
-
Autosomal dominant
- AR:
-
Autosomal recessive
- MRI:
-
Magnetic resonance imaging
- SPG4:
-
Spastic paraplegia type 4
- KSPD:
-
Kyoto Scale of Psychological Development
- EEG:
-
Electroencephalography
References
Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6(1):65–76. https://doi.org/10.1007/s11910-996-0011-1.
Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1(8334):1151–5. https://doi.org/10.1016/s0140-6736(83)92879-9.
Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev Neurol (Paris). 2015;171(6–7):505–30. https://doi.org/10.1016/j.neurol.2015.02.017.
Solowska JM, Baas PW. Hereditary spastic paraplegia SPG4: what is known and not known about the disease. Brain. 2015;138(Pt 9):2471–84. https://doi.org/10.1093/brain/awv178.
Ogasawara M, Saito T, Koshimizu E, Akasaka N, Sasaki M. A p.Arg499His Mutation in SPAST Is Associated with Infantile Onset Ascending Spastic Paralysis Complicated with Dysarthria and Anarthria. Neuropediatrics. 2019;50(6):391–4. https://doi.org/10.1055/s-0039-1694973.
Polymeris AA, Tessa A, Anagnostopoulou K, Rubegni A, Galatolo D, Dinopoulos A, et al. A series of Greek children with pure hereditary spastic paraplegia: clinical features and genetic findings. J Neurol. 2016;263(8):1604–11. https://doi.org/10.1007/s00415-016-8179-z.
Gillespie MK, Humphreys P, McMillan HJ, Boycott KM. Association of Early-Onset Spasticity and Risk for cognitive impairment with mutations at amino acid 499 in SPAST. J Child Neurol. 2018;33(5):329–32. https://doi.org/10.1177/0883073818756680.
Schieving JH, de Bot ST, van de Pol LA, Wolf NI, Brilstra EH, Frints SG, et al. De novo SPAST mutations may cause a complex SPG4 phenotype. Brain. 2019;142(7):e31. https://doi.org/10.1093/brain/awz140.
Crippa F, Panzeri C, Martinuzzi A, Arnoldi A, Redaelli F, Tonelli A, et al. Eight novel mutations in SPG4 in a large sample of patients with hereditary spastic paraplegia. Arch Neurol. 2006;63(5):750–5. https://doi.org/10.1001/archneur.63.5.750.
Shoukier M, Neesen J, Sauter SM, Argyriou L, Doerwald N, Pantakani DV, et al. Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur J Hum Genet. 2009;17(2):187–94. https://doi.org/10.1038/ejhg.2008.147.
Takezawa Y, Kikuchi A, Haginoya K, Niihori T, Numata-Uematsu Y, Inui T, et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann Clin Transl Neurol. 2018;5(5):538–51. https://doi.org/10.1002/acn3.551.
Sakaguchi Y, Uehara T, Sasaki M, Fujimura K, Kishi K, Kosaki K, et al. Hereditary spastic paraplegia masqueraded by congenital melanocytic nevus syndrome: Dual pathogenesis of germline non-mosaicism and somatic mosaicism. Eur J Med Genet. 2020;63(4):103803. https://doi.org/10.1016/j.ejmg.2019.103803.
de Souza PV, Bortholin T, Naylor FG, de Rezende Pinto WB, Oliveira AS. Infantile-onset ascending spastic paraplegia phenotype associated with SPAST mutation. J Neurol Sci. 2016;371:34–5. https://doi.org/10.1016/j.jns.2016.10.017.
Zech M, Jech R, Boesch S, Skorvanek M, Weber S, Wagner M, et al. Monogenic variants in dystonia: an exome-wide sequencing study. Lancet Neurol. 2020;19(11):908–18. https://doi.org/10.1016/S1474-4422(20)30312-4.
Xiong J, Chen S, Pang N, Deng X, Yang L, He F, et al. Neurological diseases with autism Spectrum disorder: role of ASD risk genes. Front Neurosci. 2019;13:349. https://doi.org/10.3389/fnins.2019.00349.
Park SY, Ki CS, Kim HJ, Kim JW, Sung DH, Kim BJ, et al. Mutation analysis of SPG4 and SPG3A genes and its implication in molecular diagnosis of Korean patients with hereditary spastic paraplegia. Arch Neurol. 2005;62(7):1118–21. https://doi.org/10.1001/archneur.62.7.1118.
van de Pol LA, Vermeulen RJ, van ’t Westende C, van Schie PEM, Bolster EAM, van Ouwerkerk P, et al. Risk Factors for Dystonia after Selective Dorsal Rhizotomy in Nonwalking Children and Adolescents with Bilateral Spasticity. Neuropediatrics. 2018;49(1):44–50. https://doi.org/10.1055/s-0037-1607395.
Depienne C, Tallaksen C, Lephay JY, Bricka B, Poea-Guyon S, Fontaine B, et al. Spastin mutations are frequent in sporadic spastic paraparesis and their spectrum is different from that observed in familial cases. J Med Genet. 2006;43(3):259–65. https://doi.org/10.1136/jmg.2005.035311.
Kim TH, Lee JH, Park YE, Shin JH, Nam TS, Kim HS, et al. Mutation analysis of SPAST, ATL1, and REEP1 in Korean patients with hereditary spastic paraplegia. J Clin Neurol. 2014;10(3):257–61. https://doi.org/10.3988/jcn.2014.10.3.257.
Abe-Hatano C, Iida A, Kosugi S, Momozawa Y, Terao C, Ishikawa K, et al. Whole genome sequencing of 45 Japanese patients with intellectual disability. Am J Med Genet A. 2021;185(5):1468–80. https://doi.org/10.1002/ajmg.a.62138.
Acknowledgments
We wish to thank the patient and her family for participation in this study.
Funding
The following programs provide financial support for the conduct of this research, including the study design, collection, analysis, and interpretation of data, and writing of the manuscript: Grants-in-Aid from the Research Committee for Ataxic Disease (Y.T.), the Ministry of Health, Labor and Welfare, Japan, and JSPS KAKENHI Grant Number JP21K07456 (Y. T.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. The funding body did not have roles in the design of the study and data collection, analysis, and interpretation and did not play any role in writing the manuscript.
Author information
Authors and Affiliations
Contributions
HN: drafted the manuscript, performed the genetic study, and analyzed the genetic data; HS and TM: provided the clinical information; YT: supervised the entire work and revised the manuscript. All authors read and approved the contents of the case report.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The case report complies with the Helsinki Declaration, and approval was obtained from the Ethical Committee of the University of Yamanashi. Written informed consent was obtained from the parents of the patient.
Consent for publication
The parents of the patient gave written consent on behalf of the patient and the patient’s sisters for their personal and clinical details along with any identifying images to be published in this study. The patient’s father provided written informed consent for publication of this case report.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Genes known to be responsible for or associated with spinocerebellar ataxia, Charcot-Marie-Tooth disease, or HSP.
Additional file 2.
CARE-checklist.pdf.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nan, H., Shiraku, H., Mizuno, T. et al. A p.Arg499His mutation in SPAST is associated with infantile-onset complicated spastic paraplegia: a case report and review of the literature. BMC Neurol 21, 439 (2021). https://doi.org/10.1186/s12883-021-02478-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-021-02478-0