
Abstract
Background
Disease severity is tremendously variable in tuberous sclerosis complex (TSC). In contrast with the detailed guidelines available for TSC diagnosis and management, clinical practice lacks adequate tools to evaluate the prognosis, especially in the case of in utero diagnosis. In addition, the correlation between genotypes and phenotypes remains a challenge, in part due to the large number of mutations linked to TSC. In this report, we describe a case of severe TSC diagnosed in utero and associated with a specific mutation in the gene tuberous sclerosis complex 2 (TSC2).
Case presentation
A mother was referred for a thorough investigation following the observation by ultrasound of cardiac abnormalities in her fetus. The mother was healthy and reported frequent, intense and long-lasting hiccups/spasms in the fetus. The fetus of gestational age 33 weeks and 4 days was found to have multiple cardiac tumors with cardiac ultrasound. Brain magnetic resonance imaging (MRI) performed in utero revealed the presence of sub-ependymal nodules and of abnormal signals disseminated in the white matter, in the cerebral cortex and in the cerebellum. Following diagnosis of definite TSC, pregnancy interruption was chosen by the parents. Genetic testing of the fetus exposed a duplication in exon 41 of TSC2 (c.5169dupA), which was absent in the parents. The autopsy ascertained the high severity of brain damage characterized by an extensive disorganisation of white and grey matter in most cerebral lobes.
Conclusions
This case presentation is the first to depict the association between a de novo TSC2 c.5169dupA and multi-organ manifestation together with indications of a particularly high disease severity. This report can help physicians to perform early clinical diagnosis of TSC and to evaluate the prognosis.
Similar content being viewed by others

Background
Tuberous sclerosis complex (TSC) is a rare genetic disease with a reported incidence between 1/6,000 and 1/18,000 affected individuals in the population [1,2,3,4]. It can be a devastating condition and is associated with the development of benign tumors/lesions in multiple organs [5,6,7]. Tumors, which can interfere with normal organ development and/or function, are indeed generally observed in the brain, heart, skin, kidneys and lungs [6,7,8]. However, the number of organs affected as well as the severity of tumors within an organ is extremely variable [9, 10]. As a result, symptoms of the disease are enormously diverse and variable.
The predominant neurological and psychiatric manifestations of TSC include epilepsy, intellectual disability, autism, hyperactivity and mood disorders [11,12,13]. Epilepsy can manifest as infantile spasms and is present in up to 90% of TSC patients [5, 6, 9]. It has been proposed as a major factor contributing to intellectual disability [13, 14]. In parallel, sleep disturbances in children with TSC were shown to associate with poorer health [15]. These manifestations can result from the different types of specific brain lesion characteristic of TSC including subependymal nodules, cortical tubers and giant cell astrocytomas [7, 8, 16]. In general, the extent of brain lesions positively associates with neurological and psychiatric manifestations of TSC [7, 17, 18], but the severity of manifestations remains difficult to predict [9, 19]. Outside the brain, the main manifestations of TSC are skin hypomelanotic macules and angiofibromatomas, cardiac rhabdomyomas, renal angiomyolipomas, retinal hamartomas, and lung lymphangioleiomyomatomas [5, 6, 8]. However, the identification of these manifestations is limited in the context of early life, especially in cases of in utero diagnosis.
In approximately 85% of cases, TSC has been associated with genetic variations in two specific genes: tuberous sclerosis complex 1 (TSC1) and 2 (TSC2) [7, 8, 20]. Hundreds of mutations have been reported to occur in these genes in TSC, including nonsense and missense mutations, insertions and deletions [8, 9]. The type and location of TSC1 and TSC2 mutations seem to affect phenotype, but clear genotype-phenotype correlations are difficult to establish [8, 21]. In fact, specific variants are rarely associated with precise phenotypes [9, 22, 23] and with few exceptions [21, 24], most mutations have not been characterized with functional studies. Poor genotype-phenotype correlation could originate from genetic mosaicism or from the need for a second genetic event as proposed in the two-hit model [25]. Thus, genetic analysis of TSC is importantly helpful in diagnosis [8, 26], but is not necessarily informative of disease severity.
Given the very high variability in the diverse manifestations of the disease [9, 10], the diagnosis of TSC as well as the establishment of a severity prognosis remains a challenge, especially when clinical signs of the disease are observed early in life [26]. Furthermore, a major challenge remains concerning the guidance of parents with regards to a decision about pregnancy continuation when TSC signs are present before birth, even when a definite diagnosis is made, because of the absence of clear tool sets to evaluate disease severity. We here describe one case of definite TSC, diagnosed at fetal age using ultrasound and magnetic resonance imaging, and further characterized using genetic and post-mortem biometry and microscopy, which associated with multi-organ manifestations and indications of a high disease severity. Our observations can be used to guide physicians to perform early clinical diagnosis of TSC and to evaluate the prognosis.
Case presentation
Parental and clinical history
The mother was a 40-year old woman in good physical condition reporting weekly physical activity such as running and muscle workout. It was her third pregnancy with two sons of 17 and 14 years old, living and free of any disease. She had a history of gestational diabetes during her second pregnancy that required insulin supplementation, but her blood sugar was well-controlled with diet and exercise for the third pregnancy. Her blood pressure and biochemistry revealed no sign of pre-eclampsia. She was referred for an ultrasound at 33 weeks to exclude foetal brain calcification because she had visited an endemic zone for Zika virus (i.e., Cuba) at 29 weeks of gestation. Blood testing for Zika at 32 weeks of gestation was negative. The father was a healthy 41-year old male with no prior children. Both parents were non-smokers and reported no familial history of genetic or neurodevelopmental disorders.
Notably, the mother reported the feeling of frequent and intense hiccups in the fetus. More precisely, from approximately 26–27 weeks of gestation, episodes of several minutes of regularly-spaced brief spasms were experienced every day (generally one or two times per day). These episodes sometimes lasted up to 20 min and occasionally featured spasms of varied intensity.
The 33 week ultrasound performed in regular clinical setting revealed the presence of multiple cardiac tumors, indicative of rhabdomyomas and of potential TSC. The mother was then referred to CHU Ste-Justine (specialized pediatric hospital) for in depth clinical investigation. Cardiac ultrasound and magnetic resonance imaging were then performed on the same day (i.e., 33 weeks and 4 days of gestation) to investigate for TSC in the fetus.
Cardiac ultrasound
Cardiac ultrasound revealed the presence of at least 6 cardiac tumors in the fetus. One tumor was localized to the roof of the right atrium (Fig. 1a, left). It was a non-obstructive tumor of 3.04 cm circumference. At least two tumors were present in the left ventricle. The first was from the subaortic region to the apex and had a circumference of 6.26 cm (Fig. 1b, left). It appeared slightly obstructive of the subaortic region. The second was located posterior and at the apex, and had a circumference of 3.09 cm. At least three non-obstructive tumors were present in the right ventricle. The largest (5.37 cm circumference; Fig. 1b, right) was located in the middle, the second at the apex (3.11 cm circumference), and the third at mid-ventricular position near the septum (3.39 cm circumference). These observations are strongly indicative of multiple cardiac rhabdomyomas.

Fetal cardiac ultrasound showing the presence of tumors. a Image showing the two atriums and two ventricles with the presence of tumors (T) in the right atrium (observable on the left), the right ventricle as well as in the left ventricle (observables on the right). b Image showing tumors (T) observed in the left and right ventricles. The largest tumor (6.26 cm circumference), which was observed in the subaortic region of the left ventricle and was slightly obstructive, is surrounded by a dotted line
The left and right atriums were of normal size with the membrane of the oval foramen curving towards the left atrium. The left and right ventricles were also of normal size with atrioventricular and ventriculoarterial concordances. The mitral and tricuspid valves appeared normal with normal flux. The cardiac rhythm was regular, with no sign of arrhythmia. The left and right pulmonary veins appeared normally connected to the left atrium, and caval veins to the right atrium. The umbilical vein was of normal size with normal implantation of ductus venosus. The aortic valve (5.4 mm) and pulmonary valve (6.5 mm) also appeared normal. The aortic arc and isthmus appeared of good size.
Blood circulation was observed to be relatively normal. More precisely, normal flux was measured at the level of pulmonary veins and ductus venosus (S: 89 cm/s, A: 33 cm/s, pulsatility index [PI]: 0.78), at the level of mitral and tricuspid valves, as well as at the level of ascending aorta, pulmonary arteria, and arterial canal. There was a slight acceleration of the left ventricle-aorta axis (100 cm/s), likely resulting from the proximity of one of the largest tumors. Lastly, the resistance index of uterine arteries was normal (right: 0.45, left: 0.4, late systolic notch [LSN]: 0.59), and velocimetry was also normal for umbilical arteries both at the abdominal insertion (PI: 0.98, LSN: 1.3) and the placental insertion (PI: 1.13, LSN: 1.2).
Magnetic resonance imaging
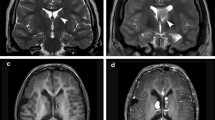
Magnetic resonance imaging (MRI) was performed the same day as the cardiac ultrasound to screen for additional markers of TSC in the fetus, with a particular focus on the central nervous system. Multiple and well-defined subependymal nodules were observed at the surrounding of both the left and right lateral brain ventricles (Fig. 2a to c). In addition, regions of abnormal signal were detected in the white matter (Fig. 2a and c) and several hyper-intense regions indicative of cortical tubers were observed throughout the cerebral cortex (Fig. 2b and c). A hyper-intense region was also localized to the left cerebellum and the possibility of cysts to kidneys was also raised.

Fetal brain MRI showing nodules and hyper-intense regions, and nucleotide call from sequencing. a Coronal T2 image showing the presence of a sub-ependymal nodule (yellow arrow, also in b and c) and an arc-shaped white matter signal abnormality (white arrow, also in c). b Axial T2 image showing the presence sub-ependymal nodules and of a hyper-intense region (red arrow, also in c). c Axial T2 image showing a sub-ependymal nodule, a white matter signal abnormality and two hyper-intense regions compatible with the presence of cortical tubers. d Chromatogram and nucleotide call shown for the forward and reverse sequencing of the mutated allele only (DNA obtained from amniotic liquid) for nucleotides 5164 to 5175 of the coding sequence of TSC2 (ENST00000219476.9). The A duplication after position 5169 is highlighted by the red box. e Sanger sequencing chromatogram and nucleotide call for the forward strand of the DNA amplicon obtained from the father sperm shown for TSC2 nucleotides 5164 to 5175 of the coding sequence. No sign of A duplication was observed. f Sanger sequencing chromatogram and nucleotide call for the forward strand of the DNA amplicon obtained from the mother skin biopsy shown for TSC2 nucleotides 5164 to 5175 of the coding sequence. No sign of A duplication was found
Together, observations from the cardiac ultrasound and the MRI confirmed the diagnosis of TSC in the fetus given the finding of two of the major criteria for the disease (i.e., cardiac rhabdomyomas and subependymal nodules). The definite TSC diagnosis was further supported by additional signs of TSC (e.g., hyper-intense cortical and cerebellar regions compatible with tubers). At this stage, medical pregnancy interruption was chosen by the parents. The interruption was executed at 34 weeks and 3 days of gestation together with an amniocentesis in order to perform genetic screening (see below). The parents also requested an autopsy of the fetus, of which the main findings are reported hereafter.
Genetic screening
The definite diagnosis of TSC strongly suggested the presence of a genetic modification in one of the two genes commonly associated with the disease, namely TSC1 and TSC2 [7, 9, 20]. The TSC1 gene product is the protein hamartin and that of TSC2 is tuberin, both of which are involved in the protein synthesis pathway [8, 27]. In general, the disease symptomatology is more severe in patients with mutations in TSC2 compared to TSC1 [8, 20, 21, 28]. A sequencing analysis of the TSC1 and TSC2 genes, including a search for duplication and deletion, was performed on the amniotic liquid sample by GeneDx (Gaithersburg, MD).
Genetic screening revealed that the fetus carried one copy (heterozygous) of a duplication of an adenine (A) at position 5169 of the coding sequence of the TSC2 gene (TSC2 c.5169dupA; Fig. 2d). According to genomic sequence identification number NG_005895.1 (NCBI) and transcript identification number ENST00000219476.9 (ensembl.org), this duplication occurs in exon 41 of the TSC2 gene that contains 42 exons. The TSC2 c.5169dupA modification is predicted to cause a frameshift starting with the codon 1724 coding for a glutamine and resulting in a change of this amino acid into a threonine and in a premature stop codon at position 5 after the reading frame is mutated (therefore denoted p.Gln1724ThrfsX5). This duplication is thus expected to be associated with nonsense-mediated mRNA decay and loss of function (or alternatively with abnormal protein function resulting from a mutated and truncated protein of 1727 amino acids instead of 1807 for the intact tuberin). Given that the c.5169dupA variant of TSC2 is not observed in large population cohorts [29], it is considered a pathogenic variant. However, although it is recognized as being pathogenic in clinical setting, to our knowledge, no study to date has reported a causal relationship between TSC2 c.5169dupA and TSC. No other TSC1 and TSC2 gene variant was detected by sequencing and deletion/duplication analysis of the fetal sample.
To verify if the genetic variant was a de novo mutation, as is the case for over two-thirds of TSC cases [8, 20], the blood of both parents was screened for the specific mutation identified in the fetus. The parents showed an absence of detectable TSC2 c.5169dupA mutation in their blood sample. The presence of this specific mutation in the germline, specifically in paternal gametes, was further screened using DNA extraction, PCR amplification and Sanger sequencing of a sperm sample of the father (with forward primer 5′-GTTCTGGCGTGACCACCAAGTC-3′, and reverse primer 5′- CAATCTGTGCAGGGGCTTTGCTA-3′). This last genetic test revealed an absence of c.5169dupA (Fig. 2e). Given that hypomelanotic macules in TSC manifest as white skin spots, genetic testing was also performed on a biopsy of a small white skin spot (3 mm2) of the mother, which also revealed an absence of the fetal mutation (Fig. 2f). The absence of the fetal mutation in the blood and tissues of the parents thus supports a de novo mutation.
Post-mortem biometry and histology
The autopsy revealed a fetus of 2556.2 g (90th percentile for 34 weeks of gestation) and 50 cm (> 90th percentile), and of 34 cm cranio-caudal length (> 90th percentile). The head circumference was 30 cm, the thorax circumference 28 cm, and that of the abdomen 24 cm. In general, biometric measurements corresponded to normal values for a gestational age of 37 weeks. The fetus did not present signs of dysmorphic features (e.g., normal ears, neck, nasal and buccal cavities, normally developed limbs). In general, the internal position and configuration of organs also appeared normal. More precisely, the left lung presented two lobes and was 24 g; the right lung presented three lobes and was 30 g; lung tissue appeared visually normal; the digestive track also appeared normal, with the liver weight of 140 g and pancreas 2.4 g. Similarly, macroscopic evaluation of the kidney, bladder and ureters revealed no abnormal position or configuration (left kidney: 14.3 g, right kidney: 14.1 g, left adrenal gland 4.5 g, right adrenal gland: 5.1 g). The same applied for the thymus, thyroid, spleen, as well as male genital organs. The results of the cardiac ultrasound were confirmed by the observation of multiple tumors in the heart.
The brain weight post-fixation (formalin zinc) was 380 g. The fronto-occipital diameters were about 12.1 cm. Brain biometric measurements were corresponding to a gestational age of 37–38 weeks and were higher than the 95th percentile for a 34-week fetus. The examination of the surface of cerebral hemispheres revealed the normal presence of all primary sulci. However, some gyri appeared to have a pachygyric aspect, with a firm consistency upon touch suggesting the presence of cortical tubers (Fig. 3a). These firm areas were noted to be predominant in the left hemisphere, particularly in the left frontal lobe. The external morphology of the spinal cord appeared normal.

Cortical tuber and sub-ependymal nodule images and histology. a Widening of a gyrus caused by a cortical tuber. b Section of the cortical tuber showing a disorganized architecture of the cerebral cortex and the blurred margin between the grey and white matter. c Histologically, the cortical tuber is characterized by large abnormal astrocytic cells, sometimes with multiple nuclei, and by round “balloon cells” with a peripheral nucleus. d Large subependymal nodule (indicated by arrows) bulging in the ventricle. e Subependymal nodule located in the germinal zone, close to the head of the caudate. f Histologically, the subependymal nodule is made up of a mixture of elongated glial cells and some “balloon cells”
The examination of coronal slices of cerebral hemispheres revealed areas of firmer consistency in the white matter and at the hemispheric surface. Microscopic examination exposed the presence of diffuse lesions of the white matter composed of globular astrocytes and organized in track alongside the axis of vessels. These lesions were associated with typical “balloon cells” and were abundant and extended, appearing in almost all cerebral lobes and sometimes almost occupying an entire gyrus. When they involved the cerebral cortex, they were responsible for a disorganization of the cortical cytoarchitecture (Fig. 3b). Small and numerous cortical lesions (tubers) were observed at the surface of the cortex, sometimes isolated but most often in continuity with white matter lesions described above (Fig. 3a and b). These were characterized by large abnormal astrocytic cells and round “balloon cells” (Fig. 3c). Astrocytes of white matter lesions and tubers strongly expressed the protein GFAP (glial fibrillary acidic protein), which was not the case for nearby balloon cells. These lesions were also accompanied with microglial activation (sometimes particularly marked). Brain ventricles did not appear to be enlarged but numerous bilateral subependymal nodules were observed in lateral ventricles (Fig. 3d). Nodules were found in the germinal zone, near the sulcus terminalis, in anterior, near the head of the caudate nucleus (Fig. 3e), and in posterior, close to the thalamus. They were made up of large astrocytes with elongated cytoplasm (Fig. 3f), showing a weak GFAP staining. The examination of axial slices of the mesencephalon showed a very narrow aqueduct of Sylvius. One small lesion, suggestive of a tuber, was also found in the cerebellum, of which the parenchyma was intensely congested. No calcification was observed at this prenatal/precocious stage. The multiplicity and extent of cortical lesions and subependymal nodules observed in the fetus supported a particularly severe form of TSC.
Discussion and conclusions
In this report, a particularly severe case of TSC was associated with a pathogenic mutation in the TSC2 gene. Firstly, a definite diagnosis of TSC was made in utero with observations of multiple cardiac rhabdomyomas and subependymal nodules using cardiac ultrasound and MRI at gestational age 33 weeks. Secondly, genetic testing revealed a duplication in exon 41 of the gene coding for the protein tuberin, predicted to change and truncate the reading frame. Thirdly, a high severity of brain damage was established with brain histology revealing widespread lesions in both the white and grey matter of the cerebrum together with multiple subependymal nodules. Together, these observations stress the importance of multi-organ investigation in prenatal diagnosis of TSC, support the literature linking TSC2 mutations to a higher disease severity, and can assist the establishment of a prognosis and of a treatment strategy in the context of prenatal TSC diagnosis.
The present observations highlight the importance of a third trimester (here 33 weeks) ultrasound during pregnancy to detect neurodevelopmental diseases. A third trimester ultrasound investigation is rarely implemented as a routine examination in clinical settings although it has a definite value for early diagnosis and intervention [30], especially in the context of TSC [26, 31]. Nonetheless, in some countries, this specific examination seems more common given the relatively frequent occurrence of TSC diagnosis in utero [31]. An early diagnosis of TSC is critical for the establishment of both an optimal monitoring (comprising regular electroencephalography) and a treatment strategy [31], which can benefit cognitive outcomes when implemented before seizure onset [32]. In the present severe case, the choice of medical pregnancy interruption, which is legal in Canada, was permitted by the third trimester ultrasound.
In addition, this report provides support for the systematic use of investigation with multiple modalities in the context of fetal diagnosis of TSC, in particular concerning the combination of ultrasound and MRI (and of genetics). This is in line with recommendations made by others with regards to the value of multi-organ investigation in TSC and that of fetal MRI [26, 31, 33]. Moreover, using this combined set of tools may also eventually serve to inform parents of the (high) likelihood of severe disease. Even if it remains unclear whether TSC severity is higher when brain lesions are observed in utero [26], in the present case, the simultaneous observation of multiple features of the disease (many large rhabdomyomas, numerous bilateral subependymal nodules, abnormal white matter signal, hyper-intense cortical and cerebellar regions) not only enabled a definite diagnosis but also suggested a more severe form of the disease. Given that the extent of brain lesions in TSC generally correlates with neurological and psychiatric symptoms [7, 17, 18, 34], and that it is also the case for lesions in other organs such as cardiac rhabdomyomas [5], inference on prognosis can (and should) be made. In the future, refinement of fetal MRI may also allow improved quantitative and qualitative measurements of cortical lesions that will importantly inform prognosis given that, for instance, global tuber load and the presence of cyst-like tubers can be indicative of more severe infantile spasms and epilepsy [34, 35].
The high severity of the present TSC case was also strongly supported by genetic testing and histology. Indeed, the identification of a duplication in the TSC2 gene in the fetus having multiple and large lesions is in agreement with a more severe symptomatology generally observed in patients with TSC2 mutations in comparison to patients with TSC1 mutations or no identified mutation [8, 9, 20, 21, 28]. The fact that the parents did not show the presence of the mutation is also supportive of a more severe disease in cases of sporadic TSC2 mutations [9]. As performed for other genetic variants related to TSC [21, 24], functional studies are now required to investigate whether TSC2 c.5169dupA leads to an altered mRNA processing, an abnormal protein function or a loss of function. TSC2 c.5169dupA is not listed in the Leiden Open Variation Database (LOVD, http://www.lovd.nl/TSC2) [36] and is therefore a novel mutation, similar to those described in a recent case report [37]. However, c.5169dupA generates a modification of Gln1724, which is reported a few times in the LOVD. Together, these observations point to a key genomic region of TSC2 in the context of TSC. Given the potential for severity of TSC, even if precise TSC1 and TSC2 mutations are rarely associated with well-defined phenotypes [9, 22, 23], there is a need to systematically inform future parents about the disease and to offer early pregnancy genetic testing (at least for pathological mutations that have been associated to severe TSC), similar to what is currently offered for Down syndrome.
Infantile spasms cannot be ascertained during the fetal period due to the impossibility to record the fetus electroencephalogram. Nevertheless, given the association between early onset epilepsy/infantile spasms and disease severity [23, 27], developing tools to investigate early signs of infantile spasms during pregnancy is likely important to complement the evaluation of disease severity and to better inform parents. In the present case, the mother had reported a potential additional sign of TSC during pregnancy, in the presence of frequent and intense spasm episodes in the fetus described as a hiccup sensation. Although there is actually no evidence for this particular report to be indicative of infantile spasms, more attention to this type of anecdotal observation and its association with cardiac and brain lesions in the context of the diagnosis of TSC during pregnancy seems relevant. In fact, clinical research is required to determine whether the interrogation of the mother about fetal hiccups/spasms may assist TSC diagnosis or the evaluation of TSC severity.
In general, better education of the population and especially of future mothers about this rare genetic disease is important to minimize psychological distress upon TSC diagnosis in the progeny. The present report emphasizes the importance of a detailed multi-organ investigation accompanied by genetic testing in the context of TSC diagnosis during the fetal period. It also supports the evaluation of TSC severity using the same tool set. Severity assessment is tremendously important in guiding the parents in the context of medical pregnancy interruption as well as regarding the suitability of early treatment. The use of mTOR inhibitors is generally well-tolerated in patients, including infants, and has been associated with clinical improvements in young TSC patients with different types of tumors [38,39,40,41]. For instance, everolimus was shown to reduce the size of cardiac tumors and brain abnormalities in neonates [40, 41], and could be used in similar cases of apparent severe TSC. Such cases could also benefit from early interventions with antiepileptic drugs [32] and even from treatment of the mother in the course of pregnancy after TSC diagnosis in utero, in order to prevent/diminish some manifestations of the disease.
Availability of data and materials
The raw acquisitions used and analysed in the context of the current Case report are available from corresponding authors on reasonable request.
Abbreviations
- CHU:
-
Centre hospitalier universitaire
- GFAP:
-
Glial fibrillary acidic protein
- LSN:
-
Late systolic notch
- MRI:
-
Magnetic resonance imaging
- mRNA:
-
Messenger ribonucleic acid
- PI:
-
Pulsatility index
- TSC:
-
Tuberous sclerosis complex
- TSC1 :
-
Tuberous sclerosis complex 1
- TSC2 :
-
Tuberous sclerosis complex 2
References
Ahlsén G, Gillberg IC, Lindblom R, Gillberg C. Tuberous sclerosis in Western Sweden. A population study of cases with early childhood onset. Arch Neurol. 1994;51(1):76–81.
Kristof AS, Zhi Li P, Major P, Landry JS. Lymphangioleiomyomatosis and tuberous sclerosis complex in Quebec: prevalence and health-care utilization. Chest. 2015;148(2):444–9.
Welin KO, Carlqvist P, Svensson A, Althin R, Eklund E, Rask O. Epilepsy in tuberous sclerosis patients in Sweden - healthcare utilization, treatment, morbidity, and mortality using national register data. Seizure. 2017;53:4–9.
Ebrahimi-Fakhari D, Mann LL, Poryo M, Graf N, von Kries R, Heinrich B, et al. Incidence of tuberous sclerosis and age at first diagnosis: new data and emerging trends from a national, prospective surveillance study. Orphanet J Rare Dis. 2018;13(1):117.
Jeong A, Wong M. Systemic disease manifestations associated with epilepsy in tuberous sclerosis complex. Epilepsia. 2016;57(9):1443–9.
Wilbur C, Sanguansermsri C, Chable H, Anghelina M, Peinhof S, Anderson K, et al. Manifestations of tuberous sclerosis complex: the experience of a provincial clinic. Can J Neurol Sci. 2017;44(1):35–43.
Nelson SL, Wild BM. Tuberous sclerosis complex: early diagnosis in infants. Pediatr Neurol Briefs. 2018;32:12.
Rosset C, Netto CBO, Ashton-Prolla P. TSC1 and TSC2 gene mutations and their implications for treatment in tuberous sclerosis complex: a review. Genet Mol Biol. 2017;40(1):69–79.
Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80.
Kingswood C, Bolton P, Crawford P, Harland C, Johnson SR, Sampson JR, et al. The clinical profile of tuberous sclerosis complex (TSC) in the United Kingdom: a retrospective cohort study in the clinical practice research Datalink (CPRD). Eur J Paediatr Neurol. 2016;20(2):296–308.
de Vries PJ, Hunt A, Bolton PF. The psychopathologies of children and adolescents with tuberous sclerosis complex (TSC): a postal survey of UK families. Eur Child Adolesc Psychiatry. 2007;16(1):16–24.
Muzykewicz DA, Newberry P, Danforth N, Halpern EF, Thiele EA. Psychiatric comorbid conditions in a clinic population of 241 patients with tuberous sclerosis complex. Epilepsy Behav. 2007;11(4):506–13.
Benova B, Petrak B, Kyncl M, Jezdik P, Maulisova A, Jahodova A, et al. Early predictors of clinical and mental outcome in tuberous sclerosis complex: a prospective study. Eur J Paediatr Neurol. 2018;22(4):632–41.
Wang YY, Pang LY, Ma SF, Zhang MN, Liu LY, Zou LP. Epilepsy may be the major risk factor of mental retardation in children with tuberous sclerosis: a retrospective cohort study. Epilepsy Behav. 2017;77:13–8.
Trickett J, Heald M, Oliver C, Richards C. A cross-syndrome cohort comparison of sleep disturbance in children with smith-Magenis syndrome, Angelman syndrome, autism spectrum disorder and tuberous sclerosis complex. J Neurodev Disord. 2018;10(1):9.
O'Callaghan FJ, Martyn CN, Renowden S, Noakes M, Presdee D, Osborne JP. Subependymal nodules, giant cell astrocytomas and the tuberous sclerosis complex: a population-based study. Arch Dis Child. 2008;93(9):751–4.
Bolton PF, Clifford M, Tye C, Maclean C, Humphrey A, le Maréchal K, et al. Intellectual abilities in tuberous sclerosis complex: risk factors and correlates from the tuberous sclerosis 2000 study. Psychol Med. 2015;45(11):2321–31.
Mous SE, Overwater IE, Vidal Gato R, Duvekot J, Ten Hoopen LW, Lequin MH, et al. Cortical dysplasia and autistic trait severity in children with tuberous sclerosis complex: a clinical epidemiological study. Eur Child Adolesc Psychiatry. 2018;27(6):753–65.
Kaczorowska M, Jurkiewicz E, Domańska-Pakieła D, Syczewska M, Lojszczyk B, Chmielewski D, et al. Cerebral tuber count and its impact on mental outcome of patients with tuberous sclerosis complex. Epilepsia. 2011;52(1):22–7.
Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9(2):88–100.
Overwater IE, Swenker R, van der Ende EL, Hanemaayer KB, Hoogeveen-Westerveld M, van Eeghen AM, et al. Genotype and brain pathology phenotype in children with tuberous sclerosis complex. Eur J Hum Genet. 2016;24(12):1688–95.
Humphrey A, Higgins JN, Yates JR, Bolton PF. Monozygotic twins with tuberous sclerosis discordant for the severity of developmental deficits. Neurology. 2004;62(5):795–8.
Curatolo P, Moavero R, Roberto D, Graziola F. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22(4):259–73.
Dufner Almeida LG, Nanhoe S, Zonta A, Hosseinzadeh M, Kom-Gortat R, Elfferich P, et al. Comparison of the functional and structural characteristics of rare TSC2 variants with clinical and genetic findings. Hum Mutat. 2020;41(4):759–73.
Jozwiak J, Jozwiak S, Wlodarski P. Possible mechanisms of disease development in tuberous sclerosis. Lancet Oncol. 2008;9(1):73–9.
Dragoumi P, O'Callaghan F, Zafeiriou DI. Diagnosis of tuberous sclerosis complex in the fetus. Eur J Paediatr Neurol. 2018;22(6):1027–34.
Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Genetic etiologies, diagnosis, and treatment of tuberous sclerosis complex. Annu Rev Genomics Hum Genet. 2019;20:217–40.
Kothare SV, Singh K, Chalifoux JR, Staley BA, Weiner HL, Menzer K, et al. Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia. 2014;55(7):1025–9.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome aggregation consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Ray CL, Grangé G. Routine third trimester ultrasound in low risk pregnancy confers no benefit!: AGAINST: Arguments for a routine third trimester ultrasound: what the meta-analysis does not show! BJOG. 2016;123(7):1122.
Słowińska M, Jóźwiak S, Peron A, Borkowska J, Chmielewski D, Sadowski K, et al. Early diagnosis of tuberous sclerosis complex: a race against time. How to make the diagnosis before seizures? Orphanet J Rare Dis. 2018;13(1):25.
Jóźwiak S, Kotulska K, Domańska-Pakieła D, Lojszczyk B, Syczewska M, Chmielewski D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15(5):424–31.
Goel R, Aggarwal N, Lemmon ME, Bosemani T. Fetal and maternal manifestations of tuberous sclerosis complex: value of fetal MRI. Neuroradiol J. 2016;29(1):57–60.
Jesmanas S, Norvainytė K, Gleiznienė R, Šimoliūnienė R, Endzinienė M. Different MRI-defined tuber types in tuberous sclerosis complex: quantitative evaluation and association with disease manifestations. Brain Dev. 2018;40(3):196–204.
Chu-Shore CJ, Major P, Montenegro M, Thiele E. Cyst-like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology. 2009;72(13):1165–9.
Leiden Open Variation Database (LOVD), Leiden University Medical Center, Leiden, The Netherlands, 2004–2020. http://www.lovd.nl/TSC2. Accessed Dec 2019.
Lee JM, Kim JH, Choi SH. Tuberous sclerosis complex case series with novel duplication mutations of TSC2. Clin Neurol Neurosurg. 2019;186:105498.
Aw F, Goyer I, Raboisson MJ, Boutin C, Major P, Dahdah N. Accelerated cardiac rhabdomyoma regression with everolimus in infants with tuberous sclerosis complex. Pediatr Cardiol. 2017;38(2):394–400.
Birca A, Mercier C, Major P. Rapamycin as an alternative to surgical treatment of subependymal giant cell astrocytomas in a patient with tuberous sclerosis complex. J Neurosurg Pediatr. 2010;6(4):381–4.
Goyer I, Dahdah N, Major P. Use of mTOR inhibitor everolimus in three neonates for treatment of tumors associated with tuberous sclerosis complex. Pediatr Neurol. 2015;52(4):450–3.
Mlczoch E, Hanslik A, Luckner D, Kitzmüller E, Prayer D, Michel-Behnke I. Prenatal diagnosis of giant cardiac rhabdomyoma in tuberous sclerosis complex: a new therapeutic option with everolimus. Ultrasound Obstet Gynecol. 2015;45(5):618–21.
Acknowledgements
Authors are thankful to the family for accepting to share information on clinical investigations, and to the teams of the CIDP and GARE of the CHU Ste-Justine, in particular to Dr. Marie-Hélène Iglesias, Dr. Sébastien Perreault, Dr. Isabelle Boucoiran, Dr. Maftei and Sophie Brisebois. Authors also thank Erika Bélanger-Nelson for a critical review of the text and language revision, and Sarah Waltho from GeneDx and Lydia Hannou for technical assistance. This case report was performed in memory of an angel.
Funding
This work was supported by the Canada Research Chair in Sleep Molecular Physiology and the Government of Québec (Canada). The funders had no role in any step of the study design, data collection and interpretation, or in manuscript preparation and submission.
Author information
Authors and Affiliations
Contributions
VM and PM assembled the clinical data and revised the manuscript. VM wrote the first version of the manuscript. NHVD analysed cardiac ultrasound data and provided images. FR analysed MRI data and provided images. CFB performed histopathological analyses and provided microscopy images. PM, JM and LC accompanied the family during diagnosis and follow-up, and contributed to data interpretation. JDG performed skin and sperm DNA extraction, amplification and analysis. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The collection of the data was conducted in the context of clinical practice in accordance with ethical standards of the Ministry of Health and Social Services of the Government of Québec (Canada). Parents provided consent to the Case report presentation.
Consent for publication
Written informed consent was obtained from the parents for publication of this Case report and of all accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mongrain, V., van Doesburg, N.H., Rypens, F. et al. A case report of severe tuberous sclerosis complex detected in utero and linked to a novel duplication in the TSC2 gene. BMC Neurol 20, 324 (2020). https://doi.org/10.1186/s12883-020-01905-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-020-01905-y