
Abstract
Background
As little is known of association(s) between gut microbiota profiles and host immunological markers, we explored these in children with and without multiple sclerosis (MS).
Methods
Children ≤18 years provided stool and blood. MS cases were within 2-years of onset. Fecal 16S rRNA gene profiles were generated on an Illumina Miseq platform. Peripheral blood mononuclear cells were isolated, and Treg (CD4+CD25hiCD127lowFoxP3+) frequency and CD4+ T-cell intracellular cytokine production evaluated by flow cytometry. Associations between microbiota diversity, phylum-level abundances and immune markers were explored using Pearson’s correlation and adjusted linear regression.
Results
Twenty-four children (15 relapsing-remitting, nine controls), averaging 12.6 years were included. Seven were on a disease-modifying drug (DMD) at sample collection. Although immune markers (e.g. Th2, Th17, Tregs) did not differ between cases and controls (p > 0.05), divergent gut microbiota associations occurred; richness correlated positively with Th17 for cases (r = +0.665, p = 0.018), not controls (r = −0.644, p = 0.061). Bacteroidetes inversely associated with Th17 for cases (r = −0.719, p = 0.008), not controls (r = +0.320, p = 0.401). Fusobacteria correlated with Tregs for controls (r = +0.829, p = 0.006), not cases (r = −0.069, p = 0.808).
Conclusions
Our observations motivate further exploration to understand disruption of the microbiota-immune balance so early in the MS course.
Similar content being viewed by others


Background
Multiple sclerosis (MS) is thought to be an autoimmune disease in which components of the immune system target cells in the brain and spinal cord, resulting in demyelination and axonal damage. While the cause(s) are unknown, both genetic and early life environmental exposures are implicated. Emerging studies have shown perturbations in the gut microbiota of individuals with MS relative to controls [5, 22, 27]. The gut microbiota’s role in modulating the host’s immune system could be highly relevant for MS. However, unlike the well-studied relationship between the immune system and MS disease processes, little is known of the gut microbiota-immune relationship in MS. The gut microbiota appears highly influential in stimulating a pro-inflammatory T cell response and subsequent disease in animal models of MS [4, 17]. Further, perturbations to the gut microbiota composition (dysbiosis) have been linked to other immune-mediated diseases distal from the gut, including rheumatoid arthritis, type 1 diabetes, atopic dermatitis and asthma [20]. These conditions could all have their ‘pathogenic origins’ in the immune response modulation by the microbiota [20].
Compared to adult MS, pediatric MS represents a unique opportunity to examine such associations close to the original exposures and biological onset of disease. Further, children have had a limited lifetime of exposures, and hence fewer potential confounding effects. We conducted a pilot study to explore the association(s) between the gut microbiota and blood immunological markers in disease-modifying drug (DMD) naïve and exposed pediatric MS cases early in their disease course and healthy controls.
Methods
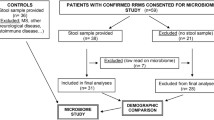
Children ≤18 years old accessing a general or MS specific pediatric clinic at the University of California, San Francisco (UCSF), USA were invited to participate in an environmental risk factor study; those providing both a stool and blood sample formed the current study cohort. MS cases had <2 years of disease (from symptom onset). Controls were similar for age and sex, were not known to have any autoimmune disorders (except for asthma or eczema) and had neither parent diagnosed with MS nor a related disorder. No child was exposed to a systemic antibiotic in the 2 months prior to stool collection. Cohort characteristics were obtained from participants via questionnaires, and standardized forms completed by the physician or research coordinator (JH) supported by chart abstraction, as described previously [27]. The UCSF Institutional Review Board approved the study. All subjects and a parent signed an assent/consent form.
Stool collection, DNA extraction and 16S rRNA profiling
Participants were asked to collect and ship overnight (on ice) a sample of the child’s first stool of the day to UCSF where it was stored at −80 °C. DNA was then extracted from approximately 25 mg of stool using PowerSoil® DNA Isolation Kit (MO BIO Laboratories, Inc, Carlsbad, CA) prior to amplification of the V4 hypervariable region of the bacterial 16S rRNA gene was amplified in triplicate [7]. Amplicons were combined, purified and pooled in equimolar concentrations and sequenced using a Miseq platform (Illumina, Inc., San Diego, CA) with a 251×151 base pair run. The 16S rRNA reads were grouped using a ≥97 % similarity threshold into operational taxonomic units (OTUs, i.e., ‘taxa’), which were singly rarefied to 201,546 reads per sample. Taxonomy was assigned using the Greengenes database via the QIIME (Quantitative Insights Into Microbial Ecology) [6, 9] platform. Community alpha diversity was expressed as richness (the number of unique taxa identified); evenness (taxon distribution), and Faith’s phylogenic diversity metric (which weights phylogenetic (i.e. evolutionary) relationships between taxa) [8], using the Vegan and Picante packages (R software [24]), Additional file 1: Appendix 1, supplementary methods.
Blood immune markers
Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient separation (Ficoll-Paque, GE Healthcare), and cryopreserved on the day of collection. Thawed PBMCs were allowed to recover in culture overnight, and then plated at 1.5 × 106 cells/ml in X-VIVO 15 media (ThermoFisher), supplemented with 5 % human AB serum (Life Technologies). Cells were stimulated with PMA (50 ng/ml) + ionomycin (0.5 μg/ml), or media alone, for 5 h in the presence of brefeldin A (GolgiPlug, BD Biosciences). Treg frequency and intracellular cytokine production by T cells were evaluated by flow cytometry. Treg (CD4+CD25hiCD127lowFoxP3+) frequency was expressed as a proportion of total CD3+ T cells. Frequencies of the following CD4+ T cell subsets were quantitated as: Th1 (IFN-γ+), Th17 (IL-17+), Th2 (IL-4+), and Tr1 (IL-10+). Total (CD3+) T cells and CD4+ T cells were reported as proportions of PBMC and total T cell populations, respectively.
Statistical analyses
Cohort characteristics were presented descriptively. Blood immune markers were compared between controls and cases (all cases, then DMD naïve and exposed separately) using non-parametric tests (Mann–Whitney and Kruskal-Wallis tests) and associations with the gut microbiota metrics (alpha diversity and phylum-level abundances) were initially explored via Pearson’s correlation coefficient. Where associations were at least ‘modest [12]’ and significant (absolute r > 0.5 and p < 0.05), linear regression analyses were performed, with the gut microbiota metrics the independent and immune markers the dependent variables, adjusting for age, disease duration, DMD exposure or case or control status, as appropriate (Additional file 1: Appendix 1). Adjusting factors were selected based on clinical relevance (i.e., disease duration, DMD exposure) and/or the limited pediatric literature which indicated that age [13] and DMD exposure [27] could influence the relationship between the developing gut microbiota and immune system. Due to the small sample size, exploration of relationships at the taxon level was not performed for this study. No corrections were applied for multiple testing, although the expected number of chance findings from the Pearson’s correlations performed were estimated as follows: seven immune markers were assessed for correlation with three diversity metrics and 13 phyla, resulting in 7×3 = 21 tests and 7×13 = 91 tests, respectively. Chance significance would be expected for one in 20 tests, i.e. approximately one test (diversity) and 4–5 tests (phylum-level). Statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS for Windows, Ver. 22.0. Armonk, NY: IBM Corp. 2013).
Results
Twenty-four children (15 relapsing-remitting MS cases, nine controls) with a mean age of 12.6 years (SD = 4.18; range 4–18) fulfilled criteria and provided both stool and blood samples; 9/24 (38 %) were boys. All cases met McDonald criteria, had relapsing-remitting MS and a short disease duration (mean = 10.0 months; range 2–23 months at stool collection). The mean age at MS onset was 11.5 years (SD = 4.84; range 4–17); seven (47 %) MS cases were exposed to a DMD and five (33 %) a corticosteroid (in the 2 months pre-stool sample). Although more controls self-identified as white (67 % controls, 33 % cases) and controls averaged almost 2 years older than cases, the proportions over 12 years were rather similar (56 % and 60 % respectively) and no characteristic (Table 1) differed significantly between cases and controls (all p > 0.2, not shown). Additional demographic, lifestyle and clinical characteristics are shown in Table 1 and Additional file 1: eTables 1.1 and 1.2, including the annualized relapse rate (mean = 0.91; SD = 0.851), time since last relapse (mean = 181.1 days; SD = 142.17) and dietary metrics (fat/fibre groupings) which were similarly distributed between cases and controls. All stool samples were collected within 3 months of the blood draw (median = 13.5 days; range 0–85 days). Most stool samples (n = 21) were collected after the blood draw, two before and one on the same day.
For the Treg analyses, 24 samples were available (n = 9 controls; n = 15 MS cases [8/15 were DMD naïve]). For the intracellular cytokine analyses, 21 samples were available (n = 9 controls; n = 12 MS cases [6/12 were DMD naïve]).
Comparison of cases and controls: gut microbiota and blood immune markers
Similar to that reported in our prior gut microbiota study [27] (from which the current cases and controls were a sub-set), there were no differences between cases (all cases as well as DMD exposed or naïve cases) and controls for any of the alpha diversity metrics (Additional file 1: eTable 2). While there were no significant differences when all cases were compared to controls for the immune markers (p > 0.05, Additional file 1: eTable 2), some differences depending on DMD exposure status were observed (Additional file 1: eTable 2). Hence, where possible, DMD exposure was considered in the analyses, either through model adjustment or a separate model developed.
Microbiota diversity-immune associations
While there were similarities for both cases and controls in the associations between their gut microbiota diversity metrics and host immunological markers, divergence was also apparent (Table 2, Fig. 1 and Additional file 1: eTable 3.1).

Divergence was observed for cases and controls in the associations between gut microbiota alpha diversity indices and peripheral blood immune markers: Gut diversity was negatively associated with Th17 in control children, but positively associated in cases. Richness is depicted for illustrative purposes. Pearson’s correlation coefficient and p-values for all children, controls, cases and by DMD exposure 
For controls, gut microbiota diversity was predominantly inversely associated with Th2 and Th17. However for cases, there were either no, or modest positive associations with Th2 and Th17, respectively (Table 2 and Fig. 1). After model adjustments, both immune markers remained significantly associated with evenness for the controls (age-adjusted p < 0.05, Additional file 1: eTable 3.1), indicating that gut microbiota dominated by specific taxa were associated with increases in these immune markers. For the cases, both richness and Faith’s diversity metric remained positively associated with Th17 (disease duration adjusted only, p = 0.008 and p = 0.013, respectively; p > 0.05 when age or DMD-adjusted, Additional file 1: eTable 3.1).
While positive associations were observed between the gut diversity metrics and Tregs, CD4+ T cells and Tr1 for both cases and controls, the strength and level of significance varied, often being more pronounced for the MS cases (Table 2). After model adjustments, significant associations remained with CD4+ T cells and Tr1 for cases only (the former when disease duration or DMD adjusted, the latter when age or disease duration but not DMD exposure adjusted). Tregs were the most strongly associated for the DMD exposed cases (evenness, r = 0.851, p = 0.015) which remained significant after age or disease duration adjustments (p = 0.034 and p = 0.017, respectively, Additional file 1: eTable 3.1).
For both cases and controls, no remarkable associations were observed between the gut microbiota diversity metrics and total T cells and Th1 (both p > 0.05; not all data shown).
Microbiota phyla-immune associations
Bacteroidetes abundance was inversely correlated with several blood immune markers for cases and controls (Table 3). Both CD4+ T cells and Tregs remained independently associated, regardless of age or DMD exposure (all adjusted p < 0.05, Additional file 1: eTable 3.2). Cases in particular exhibited strong, negative associations between Bacteroidetes abundance and immune markers such as CD4+ T cells, Tregs and Th17 (r ranged from 0.613 to 0.719, all p < 0.02). All were independent of DMD exposure, and disease duration (p < 0.02, Additional file 1: eTable 3.2). Although only Tregs remained significant after age adjustment (p = 0.042).
In contrast, positive correlations (modest or strong) were observed for controls for Th1 and Th2 (r > 0.69, p = 0.034 and p = 0.039, Table 3). However, neither remained significant after age adjustment (p > 0.05, not shown).
Actinobacteria abundance was positively associated with several blood immune markers for both cases and controls. The strongest being with Tr1 and CD4+ T cells; both were independent of age or DMD (p < 0.005, Additional file 1: eTable 3.2). However, the abundance of Fusobacteria was positively associated with Tregs for controls only (age adjusted p = 0.009, Additional file 1: eTable 3.2), with no evidence of a relationship (positive or negative) for cases (Table 3).
The Firmicutes-immune associations were mixed (Table 3); divergence between cases and controls included strong positive association with Th17 for cases only (independent of disease duration or DMD exposure, both adjusted p-values were <0.05, but not age, p = 0.089). Conversely for controls (not cases), strong inverse associations with Th1 (r = −0.789, p = 0.011) were observed, remaining significant after age adjustments (p < 0.03, Additional file 1: eTable 3.2).
Discussion
Associations were found between the composition of the gut microbiota and the host blood immune marker profiles in children with and without MS. While there were similarities for both cases and controls, there were also several divergent findings which were independent of DMD exposure. Further, these divergent microbiota-immune relationships were measurable very early in the MS disease course, as all pediatric cases were within 24 months of symptom onset. Disruption of microbiota-immune balance so early in MS is intriguing.
Gut microbiota evenness was strongly and inversely associated with Th17 and Th2 for the control children. However, there was either no or modest positive associations for the MS cases. While we were unable to find a similar study with which to compare our findings, a recent Canadian study demonstrated that diversity depleted uneven neonatal gut microbiota preceded later childhood-onset atopy and asthma risk [2]. In addition, mono-colonization i.e. an uneven community of the murine gut microbiota with segmented filamentous bacteria results in significant expansion of ileal lamina propria Th17 populations [15]. Our observations were consistent with these studies and suggest that uneven microbiota (which can indicate species overgrowth) in the pediatric gut is associated with increases in markers of Th17 and Th2 populations. Why this microbiota-immune relationship is reversed or lost in the MS children is of interest and may be due to the specific composition of gut microbial communities in MS versus control populations.
Nonetheless, there were also similarities for the cases and controls in that both groups exhibited positive associations between the gut microbiota diversity metrics and Tregs, CD4+ T cells and Tr1. Not all findings were independent of age which may be suggestive of an evolving microbiota-immune relationship as the child develops [16]. Also, although the direction of effect was similar for cases and controls, the strength of these associations were greater for the MS cases in a number of instances, particularly for the DMD exposed group. For instance, despite no significant differences between controls and cases (DMD exposed or naïve) for Tregs, this immune marker was strongly associated with diversity (evenness) for the DMD exposed cases (i.e. had a high r-value: r = 0.851, p = 0.015) which was independent of age or disease duration (p < 0.05).
Microbiota phylum abundance-immune associations
There were several microbiota-immune associations measurable at the phylum-level which were consistent across both cases and controls. These included Bacteroidetes and Actinobacteria which represent two of the most common bacterial phyla in the human gut. The relative abundance of Bacteroidetes was negatively associated with both CD4+ T cells and Tregs, and strong positive associations were observed between Actinobacteria and CD4+ T cells and Tr1, all of which were independent of age or DMD exposure. These observations concur with the wider literature. For instance, Bacteroidetes includes species such as Bacteroides fragilis, well studied in animal models for their ability to induce CD4+ T cells [20, 21]. In addition, Tr1, i.e. IL-10 are reported as one of the main immunoregulatory factors required for immune tolerance of the intestinal microbiota [20], such that a strong relationship with one of the main gut phyla would not be unexpected.
However, divergences in the phylum-immune relationship were observed. Of interest were two associations only observed in the control children; Fusobacteria abundance exhibited a strong, positive association with the Tregs and the Firmicutes phylum was inversely associated with Th1 (r = 0.829 and r = −0.789, respectively, p < 0.02). Both were independent of age. Conversely, for cases, no strong relationships (either positive or negative) were observed between these phyla and immune markers. These differences are likely due to the specific composition of these phyla, each of which consists of hundreds of functionally distinct species and strains. For example members of Clostridiales Clade IV and XIV can induce Treg cells [3] while a related Firmicute, Clostridium difficile is a well described human pathogen. Findings highlight the need to study much larger cohorts, sufficiently powered to identify specific gut microbiome members that exhibit relationships with these important adaptive immune responses.
While little is known of the gut-related Fusobacteria-immune relationships, there is some evidence to suggest that dysregulation of Tregs may contribute to disease processes in MS, specifically an increased risk of relapse [25]. Loss of the Fusobacteria-immune relationship for cases may be clinically relevant and possibly linked to our previous observation that depletion of Fusobacteria in the gut microbiota of pediatric MS cases was associated with a higher risk (hazard) of relapse [26]. It would be of value for future studies to examine the gut-immune relationship in the context of MS disease activity (i.e., clinical relapses or MRI-related metrics).
Possible mechanisms of action
While we modelled the gut microbiota as a predictor of host immune markers, the two are considered to have coevolved with likely a bi-directional relationship [18]. Although the potential mechanism(s) of action are not well understood [1], the gut microbiota are known to elicit Treg recruitment, regulate Th17 and Treg cell differentiation, which in the healthy gut promotes immune-microbiota homeostasis [19]. Metabolites produced by the gut microbiota such as short chain fatty acids (e.g., butyrate) have been shown to exert immunomodulatory affects, including down-regulation of pro-inflammatory mucosal Th2 responses [1, 10, 19, 28]. As suggested by our findings, and others, the effects of microbiota compositional changes are not exclusively local, but can influence the systemic immune response and, as others have shown, are associated with systemic inflammatory diseases distal to the gut, such as rheumatoid arthritis [19]. In the context of conditions such as inflammatory bowel disease, others have concluded that ‘dysregulated CD4 T cells responses to antigens of the microbiota leads to chronic, typically relapsing and remitting disease that reflects the immune system’s inability to eliminate the antigens that drive the abnormal response [20].’ This could apply to MS, also a predominantly relapsing-remitting disease. Evidence to suggest that gut microbiota can trigger non-gut related diseases includes the observation that Clostridia sp., Candidatus arthromitus or segmented filamentous bacteria in mice can cause inflammatory arthritis [19] and MS-like symptoms in EAE [17] mediated via Th17 production.
Strengths and limitations
Limitations of this pilot study include the small sample size and no correction for multiple testing. Some findings would be expected by chance alone, such that we have specifically focused on those that were modest or strong and significant (absolute r > 0.5 and p < 0.05), appeared consistent across related biological markers and, wherever possible, developed models adjusting for potential confounders. In addition, we purposely focused on a high-level view of the gut microbiota, i.e., diversity and phylum-level abundance. Our findings require replication, but are also necessary to help guide and power future studies. Further exploration is needed, when the sample size permits, such as: to understand the specific functions being performed by the gut microbiota, or key members of the community present; to gain a better understanding of the influence of a child’s age on the microbiota-immune relationship; and to examine the influence of host genetics [11], diet, drug exposure and other effect modifiers.
Strengths include a well-phenotyped group of children with a rare but life changing chronic condition; less than 5 % of individuals with MS develop the disease and are diagnosed during the childhood years. All MS cases were examined and diagnosed by a pediatric MS neurologist and enrolled close to symptom onset with paired stool and blood samples collected within a short time of each other. Our study offers novel insights; the gut microbiota has not been well characterized in children (e.g. the Human Microbiome Project, HMP only included adults over 18 years [14]). Further, we are not aware of another study exploring the microbiota-immune relationship in MS (adults or children).
Conclusions
Understanding the relationship between diseases considered immune-mediated, such as MS, and the gut microbiota, which has a central role in shaping the immune response, is fundamental [20]. We found associations between the gut microbiota composition and host blood immune markers which were divergent between cases and controls. Why the microbiota-immune balance appears disrupted in MS is intriguing and deserves further study. A better understanding and identification of key microbiota members involved may lead to novel opportunities to modulate immune dysregulation in diseases such as MS.
Abbreviations
- BMI:
-
Body mass index
- CD3 CD4:
-
Cluster of differentiation (protein) 3, 4
- DMD:
-
Disease-modifying drug (DMD)
- DNA:
-
Deoxyribonucleic acid
- EAE - EDSS:
-
Expanded Disability Status Scale score
- HMP:
-
Human microbiome project
- MRI:
-
Magnetic resonance imaging
- MS:
-
Multiple sclerosis (MS)
- OTU:
-
Operational taxonomic unit
- PBMC:
-
Peripheral blood mononuclear cell
- QIIME:
-
Quantitative Insights Into Microbial Ecology
- RNA:
-
Ribonucleic acid
- SD:
-
Standard deviation
- Th1 Th2:
-
Type 1, Type 2T helper (cell)
- Treg:
-
T regulatory (cell)
- UCSF:
-
University of California San Francisco
References
Ahern PP, Faith JJ, Gordon JI. Mining the human gut microbiota for effector strains that shape the immune system. Immunity. 2014;40:815–23.
Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, Kuzeljevic B, Gold MJ, Britton HM, Lefebvre DL, Subbarao P, Mandhane P, Becker A, McNagny KM, Sears MR, Kollmann T, Investigators CS, Mohn WW, Turvey SE, Brett Finlay B. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med. 2015;7:307ra152.
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–6.
Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–41.
Cantarel BL, Waubant E, Chehoud C, Kuczynski J, DeSantis TZ, Warrington J, Venkatesan A, Fraser CM, Mowry EM. Gut microbiota in multiple sclerosis: possible influence of immunomodulators. J Invest Med. 2015;63:729–34.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nature Meth. 2010;7:335–6.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME. 2012;6:1621–4.
Faith DP, Baker AM. Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol Bioinfo. 2006;2:121–8.
Faruqi AA. Microbiome Analysis using Workflow of QIIME. In: GitHub. 2015.
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–50.
Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, Spector TD, Clark AG, Ley RE. Human genetics shape the gut microbiome. Cell. 2014;159:789–99.
Hinkle DE, Wiersma W, SG J. Applied Statistics for the Behavioral Sciences. Boston: Houghton Mifflin; 2003.
Hollister EB, Riehle K, Luna RA, Weidler EM, Rubio-Gonzales M, Mistretta TA, Raza S, Doddapaneni HV, Metcalf GA, Muzny DM, Gibbs RA, Petrosino JF, Shulman RJ, Versalovic J. Structure and function of the healthy pre-adolescent pediatric gut microbiome. Microbiome. 2015;3:36.
Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98.
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jorgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clement K, Dore J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T, Meta HITc, Bork P, Wang J, Ehrlich SD, Pedersen O. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–6.
Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4615–22.
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–48.
Longman RS, Yang Y, Diehl GE, Kim SV, Littman DR. Microbiota: host interactions in mucosal homeostasis and systemic autoimmunity. Cold Spring Harbor Symp Quant Bio. 2013;78:193–201.
Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–41.
Mielcarz DW, Kasper LH. The gut microbiome in multiple sclerosis. Curr Treat Op Neurol. 2015;17:344.
Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, Chihara N, Tomita A, Sato W, Kim SW, Morita H, Hattori M, Yamamura T. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One. 2015;10, e0137429.
Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18:968–84.
R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2013.
Steinman L. Immunology of relapse and remission in multiple sclerosis. Annu Rev Immunol. 2014;32:257–81.
Tremlett H, Fadrosh DW, Faruqi AA, Hart J, Roalstad S, Graves J, Lynch S, Waubant E. Gut microbiota composition and relapse risk in pediatric MS: a pilot study. J Neurol Sci. 2016;363:153–7.
Tremlett H, Fadrosh DW, Faruqi AA, Zhu F, Hart J, Roalstad S, Graves J, Lynch S, Waubant E. Gut microbiota in early pediatric multiple sclerosis: a case–control study. Eur J Neurol. 2016. doi:10.1111/ene.13026 [Epub ahead of print].
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20:159–66.
USA’s Centers for Disease Control and Prevention. Children's BMI Tool for Schools. [updated September 13, 2010; Accessed 19 Sept 2016]; Available from: http://www.cdc.gov/healthyweight/assessing/bmi/childrens_BMI/tool_for_schools.html.
Whittaker RH. Evolution and measurement of species diversity. Taxon. 1972;21:213–51.
Acknowledgements
The US Network of Pediatric MS Centers (authors listed in alphabetical order): Greg Aaen1, Anita Belman2, Leslie Benson3, Charlie Casper4, Tanuja Chitnis3, Mark Gorman3, Yolanda Harris8, Lauren Krupp2, Tim E Lotze6, Sabeen Lulu7, Jayne Ness5, Cody Olsen4, Erik Roan4, Moses Rodriguez5, John Rose4, Timothy C Simmons4, Jan-Mendelt Tillema5, Wendy Weber4, Bianca Weinstock-Guttman9
1. Loma Linda University, Loma Linda, CA, United States; 2. Stony Brook University, Stony Brook, NY, United States; 3. Harvard University, Cambridge, MA, United States; 4. University of Utah, Salt Lake City, UT, United States; 5. Mayo Clinic, Rochester, MN, United States; 6. Baylor College of Medicine, Houston, TX, United States; 7. University of California, San Francisco, San Francisco, CA, United States; 8. University of Alabama, Birmingham, AL, United States; 9. State University of New York at Buffalo, Buffalo, NY, United States.
Funding
This work was supported in part by the National MS Society RG4861A3/1 (PI Waubant), National Institutes of Health NS071463 (PI Waubant), The Race to Erase MS (PI Waubant) and the Canada Research Chair program (PI Tremlett). The funding source(s) had no role in the study design, collection, analysis or interpretation of the data, or in the decision to submit the article for publication.
Availability of data and materials
Raw, de-identified data can be requested from the corresponding author.
Authors’ contributions
The corresponding author (HT) takes responsibility for the integrity of the data and the accuracy of the data analysis and had full access to the data. DF performed the gut microbiota analyses. CS performed the blood draws and the immunological assays. HT performed the statistical analyses. EW, JH and JG facilitated sample and data collection. SR facilitated electronic linkage and quality assurance of data from environmental risk factors parent study. All authors were involved in the current study design, contributed to interpretation of data. SL and EW designed the original study and obtained funding. DF and AF drafted the microbiota methods. CS and SZ drafted the immunology methods. HT drafted the remaining manuscript. All authors revised the manuscript and approved of the final version to be published.
Competing interests
Helen Tremlett is funded by the Canada Research Chair program. She has received research support from the National Multiple Sclerosis Society, the Canadian Institutes of Health Research, and the UK MS Trust; speaker honoraria and/or travel expenses to attend conferences from the Consortium of MS Centres (2013), the National MS Society (2012, 2014), Bayer Pharmaceuticals (2010), Teva Pharmaceuticals (2011), ECTRIMS (2011, 2012, 2013, 2014), UK MS Trust (2011), the Chesapeake Health Education Program, US Veterans Affairs (2012), Novartis Canada (2012), Biogen Idec (2014), American Academy of Neurology (2013, 2014, 2015). Unless otherwise stated, all speaker honoraria were either donated to an MS charity or to an unrestricted grant for use by her research group.
Douglas Fadrosh has no disclosures.
Ali Faruqi has no disclosures.
Janace Hart has no disclosures.
Shelly Roalstad has no disclosures.
Jennifer Graves is funded by the Race to Erase MS and the National MS Society.
Collin Spencer has no disclosures.
Susan Lynch is funded by the NIH, Sloan Foundation, Cystic Fibrosis Foundation, Broad Foundation, Jannsen Pharmaceuticals, Gilead and Pfizer. She has recently or currently acts as an ad hoc consultant for Janssen Pharmaceuticals, Regeneron, Boston Consulting Group, Theravance and Novartis. She is an Associate Editor for Microbiome, a members of the Scientific Advisory Board of Second Genome and has received honoraria for lectures from American Thoracic Society, American Academy of Allergy Asthma and Immunology, Georgia Regents University, Alta Bates and Kaiser Permanente. She holds four patents and has received royalties for IP licensed by KaloBIos Inc.
Scott Zamvil receives research grant support from the NIH (1RO1 NS092835), the NMSS (RG 4768, RG5180. RG5179), The Guthy Jackson Charitable Foundation, The Maisin Foundation, Biogen Idec, Inc. and Teva Pharmaceuticals, Inc. Currently, Dr. Zamvil serves Deputy Editor of Neurology, Neuroimmunology and Neuroinflammation and is a member of the advisory board for the International Society of Neuroimmunology. He has served as a consultant and received honoraria from Biogen-Idec, EMD-Serono, Genzyme, Novartis, Questcor, Roche, and Teva Pharmaceuticals, Inc., and has served or serves on Data Safety Monitoring Boards for Lilly, BioMS, Teva and Opexa Therapeutics
Emmanuelle Waubant is funded by the NIH, the NMSS, and the Race to Erase MS. She has received honorarium for one educational lecture from Genentech. She volunteers on an advisory board for a Novartis trial. She has received honorarium or travel support from ACTRIMS, ECTRIMS, and AAN.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The UCSF Institutional Review Board approved the study. All subjects and a parent signed an assent/consent form.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional file
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tremlett, H., Fadrosh, D.W., Faruqi, A.A. et al. Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol 16, 182 (2016). https://doi.org/10.1186/s12883-016-0703-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-016-0703-3