
Abstract
Background
LEOPARD syndrome (LS) belongs to the family of neuro-cardio-facio-cutaneous syndromes, which include Neurofibromatosis-1 (NF1), Noonan syndrome, Costello Syndrome, cardio-facio-cutaneous syndrome, Noonan-like syndrome with loose anagen hair and Legius syndrome. These conditions are caused by mutations in genes encoding proteins involved in the RAS-MAPK cellular pathway. Clinical heterogeneity and phenotype overlaps across those different syndromes is already recognized.
Case presentation
We hereby report a heterozygous de novo mutation in the PTPN11 gene (c.1403C > T) manifesting with a clinical picture of LS during childhood, and later development of neuropathic pain with hypertrophic plexi, which are typically observed in NF1 but have not been reported in LS.
Conclusion
LS caused by PTPN11 mutations may be associated with hypertrophic roots and plexi. Consequently, clinicians should be aware of the possible development of neuropathic pain and consider specific diagnostic work-up and management.
Similar content being viewed by others


Background
LEOPARD syndrome (LS) is defined by Lentigines, Electrocardiographic abnormalities, Ocular hyperthelorism, Pulmonary valve stenosis, Abnormal genitals, Retarded growth and Deafness, and is caused by heterozygous mutations in the protein tyrosine phosphatase, non-receptor type 11 gene (PTPN11). LS is a rare, autosomal dominantly inherited disease, belonging to the family of neuro-cardio-facio-cutaneous syndromes (NCFCs). These syndromes present significant genetic heterogeneity and phenotype overlaps. Here we describe a patient who arbors a heterozygous mutation in the PTPN11 gene (c.1403C > T) manifesting with a clinical phenotype of LS associated with neuropathic pain and hypertrophic roots and plexi.
Case presentation
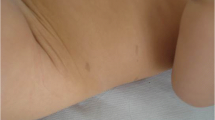
A Caucasian woman presented with multiple cutaneous diffuse lentigines and cafe-au-lait spots (7 spots of 0.7 to 5 cm of diameters, localized in the palms, feet, trunk and buttock) since the age of 6 years (Figure 1A-D), associated with mild bilateral ptosis, facial dysmorphism (such as prognathism necessitating surgery, macroglossia) and pulmonary valve stenosis. Her body weight and height was in the normal age-range and she had no psychomotor delay, working as a commercial manager. Ophthalmologic examination revealed multiple peripheral retinal lentigines. Family history was negative for neurological or cutaneous symptoms.

Clinical and MRI features of LEOPARD syndrome presenting with hypertrophic plexi and neuropathic pain. Patient with LEOPARD syndrome showing multiple cutaneous lentigines (A and C: trunk, B: palms) and café-au-lait spots, on the upper buttock (D). Spinal MRI showing hyperthrophy of the cervico-brachial (E) and lumbo-sacral (F) plexi.
She was re-examined at the age of 41 years, related to the development of progressive painful dysaesthesias. Neurological examination revealed moderate sensory-motor deficit in all four limbs, with mild facial weakness. Conventional nerve conduction studies of the upper and lower limbs were normal, but late spinal F latencies were diffusely prolonged, suggesting diffuse proximal neuropathy. Spinal MRI revealed hypertrophic cervico-brachial and lumbo-sacral plexi and roots (Figure 1E, F). Extensive diagnostic work-up excluded other inflammatory, genetic or tumor conditions associated with hypertrophic neuropathies [1–3]. MRI of the lower limbs showed two gadolinium-enhanced T2-hyperintense nodules (not shown), suggestive of neurofibromas. Sequencing of the NF1 gene showed no mutation (including search for intragenic rearrangements by MLPA analysis), while sequencing of the PTPN11 gene revealed a heterozygous missense mutation in Exon 12 (c.1403C > T, p.Thr468Met), confirming the diagnosis of LS.
Conclusion
We report a patient harboring the PTPN11 c.1403C > T mutation which manifested with a phenotype of childhood-onset LS, fulfilling clinical criteria (multiple lentigines plus 2 major features [4]), and later development of neuropathic pain related to hypertrophic nerve roots and plexi.
The association of skin changes, facial dysmorphism and mild cardiac abnormalities are almost universally present in LS patients [4]; sensorineural hearing loss and neurological deficits (neuropsychological difficulties and rarely seizures) are present in less than a third of the cases. Most abnormalities are present at birth, however some can appear during childhood, as is often the case for lentigines, or even develop during adulthood, as for ventricular hypertrophy.
Mutations in PTPN11 are responsible for 80% of LS cases. The c.1403C > T nucleotide substitution found in our patient is the most frequently reported mutation, and it leads to a decreased catalytic capacity of the SHP2 protein. RAF1 and BRAF mutations are much less frequently encountered.
LS belongs to the NCFCs family. The most prevalent disorders of this group are Noonan syndrome and Neurofibromatosis-1 (NF1), while LS is a rarer condition. NCFCs display common clinical features such as psychomotor delay, facial dysmorphism and cardiac, cutaneous and skeletal abnormalities. Although genetic causes of NCFCs are heterogeneous, the molecular mechanisms involved alter the RAS/MAPK signaling pathway [5]. RAS is a human oncogene implicated in different cellular functions and its transducing signal, involving different MAP kinases, is critical for cell proliferation and survival. Thus, its disruption may result in uncontrolled cell growth and cancer. Somatic mutations in protein components of this pathway are found in various malignancies [6]. In contrast, germ-line mutations, which result in a less dysfunctional gene product, underlie the development of NCFCs.
NCFCs display significant genetic and phenotypic heterogeneity: on the one hand, mutations in the same gene may be responsible for different syndromes, as is the case for Noonan syndrome and LS, mostly caused by mutations in the PTPN11 gene; on the other hand, dysfunction of different proteins at various levels of the RAS/MAPK cascade can lead to similar clinical presentations [7,8].
Our patient harbored a PTPN11 de novo mutation manifesting as LS associated with nerve and roots hypertrophy and neuropathic pain. Although these latter changes, associated with gadolinium-enhanced deep nodules evoking neurofibromas, are typical findings of NF1, our subject presented no NF1 gene mutations. Recently, three cases of PTPN11 mutations were reported with early cutaneous, but no neurological, features of NF1 [9]. Our patient showed cafe-au-lait spots, however without other dermatological hallmarks of NF1, such as skinfold freckling or cutaneous neurofibromas. Some authors suggested that NF1 can be misdiagnosed if based solely on the presence of these skin abnormalities, which can be encountered in other conditions and disappear with growth [10].
Our observation underscores that LS caused by PTPN11 mutations could be associated with a proximal radiculoplexopathy. This finding is of clinical relevance: while neurologic complications are common in NF1, they are not routinely screened for in LS patients. Thus, clinicians should question adult LS individuals about the development of neuropathic pain and neurological deficits, and perform careful physical examination and consider spinal MRI in search of plexi/root hypertrophy and neurofibromas.
Neurogenic pain can significantly impact quality of life, and its management may be challenging. In order to address different important aspects related to pain, especially in the context of NCFCs, a multidisciplinary approach is recommended, associating physical therapy with medical (including analgesics, opiates, antiepileptic drugs, topic anesthetics, antidepressants, or a combination) and psychological treatments [11]. For example, a multimodal approach including a relaxation response resiliency program (3RP) has been recently used in a pilot study involving patients with NF1, NF2, and schwannomatosis, with encouraging results in terms of coping strategies and quality of life [12]. The use of oral ketamine or bevacizumab has also been suggested as a therapeutic option in refractory cases of NF1 or NF2, respectively, but its efficacy only relies on single case reports [13,14]. By contrast, surgery should be considered in patients with nerve or spine compression by plexiform neurofibromas.
Additionally, oncologic screening should be considered in LS patients presenting deep nodules suggestive of neurofibromas, particularly in light of the risk for malignant transformation, in analogy to what is observed in NF1 patients [15]. This is particularly important in the perspective of promising target-based therapies acting directly on the RAS/MAPK signaling pathway, such as MEK and RAF inhibitors [16]. Although it has been suggested that these molecules may play a role in controlling tumor progression in various conditions associated with neurofibromatosis [16], their effectiveness in neurogenic pain control seems to date less prominent [17–19].
Consent
Oral informed consent was obtained from the patient for publication of this case report and any accompanying images. Since the patient was not reachable, a written consent could not be obtained.
Abbreviations
- LS:
-
LEOPARD syndrome
- PTPN11:
-
Tyrosine-protein phosphatase non-receptor type 11
- NCFCs:
-
Neuro-cardio-facio-cutaneous syndromes
- NF1:
-
Neurofibromatosis type 1
- NF2:
-
Neurofibromatosis type 2
References
Beydoun SR, Muir J, Apelian RG, Go JL, Lin FP. Clinical and imaging findings in three patients with advanced inflammatory demyelinating polyradiculoneuropathy associated with nerve root hypertrophy. J Clin Neuromuscul Dis. 2012;13:105–12.
Masuda N, Hayashi H, Tanabe H. Nerve root and sciatic trunk enlargement in Déjérine–Sottas disease: MRI appearances. Neuroradiology. 1992;35:36–7.
Heilbrun ME, Tsuruda JS, Townsend JJ, Heilbrun MP. Intraneural perineurioma of the common peroneal nerve. Case report and review of the literature. J Neurosurg. 2001;94:811–5.
Voron DA, Hatfield HH, Kalkhoff RK. Multiple lentigines syndrome. Case report and review of the literature. Am J Med. 1976;60:447–56.
Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci. 2010;1214:99–121.
De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16:S17–27.
Fryer AE, Holt PJ, Hughes HE. The cardio-facio-cutaneous (CFC) syndrome and Noonan syndrome: are they the same? Am J Med Genet. 1991;38:548–51.
De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet. 2005;77:1092–101.
Carcavilla A, Pinto I, Muñoz-Pacheco R, Barrio R, Martin-Frías M, Ezquieta B. LEOPARD syndrome (PTPN11, T468M) in three boys fulfilling neurofibromatosis type 1 clinical criteria. Eur J Pediatr. 2011;170:1069–74.
Korf BR. Diagnostic outcome in children with multiple café au lait spots. Pediatrics. 1992;90:924–7.
Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17:1113–88.
Vranceanu AM, Merker VL, Plotkin SR, Park ER. The relaxation response resiliency program (3RP) in patients with neurofibromatosis 1, neurofibromatosis 2, and schwannomatosis: results from a pilot study. J Neurooncol. 2014;120:103–9.
Soto E, Stewart DR, Mannes AJ, Ruppert SL, Baker K, Zlott D, et al. Oral ketamine in the palliative care setting: a review of the literature and case report of a patient with neurofibromatosis type 1 and glomus tumor-associated complex regional pain syndrome. Am J Hosp Palliat Care. 2012;29:308–17.
Kollár A, Hewer E, Kellner-Weldon F, Ochsenbein A. Complete pain relief after bevacizumab in a patient with neurofibromatosis type 2. Acta Oncol. 2015;54:280–3.
Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–4.
Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103–19.
Hua C, Zehou O, Ducassou S, Minard-Colin V, Hamel-Teillac D, Wolkenstein P, et al. Sirolimus improves pain in NF1 patients with severe plexiform neurofibromas. Pediatrics. 2014;133:e1792–7.
Weiss B, Widemann BC, Wolters P, Dombi E, Vinks A, Cantor A. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: a Neurofibromatosis Clinical Trials Consortium phase II study. Neuro Oncol. 2014;0:1–8. pii: nou235.
Weiss B, Widemann BC, Wolters P, Dombi E, Vinks AA, Cantor A, et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: an NF clinical trials consortium phase II study. Pediatr Blood Cancer. 2014;61:982–6.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Dr Wider receives financial support from the Leenaards Foundation.
Dr Croquelois receives financial support from the Swiss National Science Foundation.
None of the authors have non-financial competing interests to declare in relation to this manuscript.
Authors’ contributions
CW, TK and AC were involved in the clinical care of the patient, in the diagnostic process and genetic analysis. MS drafted the manuscript with the help of all the other authors. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Spatola, M., Wider, C., Kuntzer, T. et al. PTPN11 mutation manifesting as LEOPARD syndrome associated with hypertrophic plexi and neuropathic pain. BMC Neurol 15, 55 (2015). https://doi.org/10.1186/s12883-015-0310-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-015-0310-8