
Abstract
Background
Focal segmental glomerulosclerosis (FSGS), a histologic pattern of injury in the glomerulus, is one of the leading glomerular causes of end-stage renal disease (ESRD) worldwide. Despite extensive research, the underlying biological alterations causing FSGS remain poorly understood. Studying variations in gene expression profiles offers a promising approach to gaining a comprehensive understanding of FSGS molecular pathogenicity and identifying key elements as potential therapeutic targets. This work is a meta-analysis of gene expression profiles from glomerular samples of FSGS patients. The main aims of this study are to establish a consensus list of differentially expressed genes in FSGS, validate these findings, understand the disease’s pathogenicity, and identify novel therapeutic targets.
Methods
After a thorough search in the GEO database and subsequent quality control assessments, seven gene expression datasets were selected for the meta-analysis: GSE47183 (GPL14663), GSE47183 (GPL11670), GSE99340, GSE108109, GSE121233, GSE129973, and GSE104948. The random effect size method was applied to identify differentially expressed genes (meta-DEGs), which were then used to construct a regulatory network (STRING, MiRTarBase, and TRRUST) and perform various pathway enrichment analyses. The expression levels of several meta-DEGs, specifically ADAMTS1, PF4, EGR1, and EGF, known as angiogenesis regulators, were analyzed using quantitative reverse transcription polymerase chain reaction (RT-qPCR).
Results
The identified 2,898 meta-DEGs, including 665 downregulated and 669 upregulated genes, were subjected to various analyses. A co-regulatory network comprising 2,859 DEGs, 2,688 microRNAs (miRNAs), and 374 transcription factors (TFs) was constructed, and the top molecules in the network were identified based on degree centrality. Part of the pathway enrichment analysis revealed significant disruption in the angiogenesis regulatory pathways in the FSGS kidney. The RT-qPCR results confirmed an imbalance in angiogenesis pathways by demonstrating the differential expression levels of ADAMTS1 and EGR1, two key angiogenesis regulators, in the FSGS condition.
Conclusion
In addition to presenting a consensus list of differentially expressed genes in FSGS, this meta-analysis identified significant distortions in angiogenesis-related pathways and factors in the FSGS kidney. Targeting these factors may offer a viable strategy to impede the progression of FSGS.
Similar content being viewed by others

Introduction
Focal segmental glomerulosclerosis (FSGS) is a histologic pattern of injury characterized by segmental (≤ 50% of the affected glomerulus is sclerosed) and focal (≤ 50% of all glomeruli affected) sclerosis in the glomeruli [1]. Massive proteinuria and nephrotic syndrome, with a high incidence of progression to end-stage renal disease (ESRD), are the main clinical manifestations of FSGS. FSGS is classified into two main types: primary (idiopathic) and secondary. Primary FSGS is a primary glomerulopathy and a representative condition displaying nephrotic syndrome. In contrast, secondary FSGS is often associated with non-nephrotic proteinuria and exhibits less severe clinical manifestations [2,3,4]. The diagnosis of FSGS type is usually difficult due to the frequent overlaps in the clinicopathological symptoms of these two types [3, 5]. Due to significant regional and ethnic variances, determining the actual incidence and prevalence of FSGS is challenging; However, based on statistical analysis, the incidence of FSGS is about 1.4 to 21 cases per million people [6].
Despite numerous studies on FSGS, there remains an unmet need for further investigations to uncover the underlying molecular mechanisms of its pathogenesis, understand the specific features of each type, and develop novel treatments for its effective management [7]. Recently, with the advancement of high-throughput techniques and the adoption of a systems biology approach, researchers have been able to obtain a comprehensive map of small molecular changes across various disease conditions. In this context, transcriptomic data have proven particularly valuable, as they enable researchers to measure a vast number of expression variations in both healthy and diseased states [8,9,10,11]. However, the presence of false positives and contradictions among the resulting list of candidate genes is one of the primary challenges in transcriptomic data analysis. Such drawbacks may lead to a final list of differentially expressed genes (DEGs) that do not accurately represent the genes of interest in the real world [12]. False positives in transcriptomic data can arise from various factors, including elevated levels of gene-gene co-expression, the presence of highly expressed genes within specific cell types, and methodological parameters. Vigilance regarding these potential sources of error is crucial to ensuring the accuracy and reliability of transcriptomic analyses [13, 14].
Meta-analysis of transcriptomics datasets is one strategy that can provide a more consistent and reliable list of DEGs [15]. Therefore, performing a meta-analysis on several related transcriptomics datasets can significantly decrease the false positives and contradictions caused by alterations in homogeneity, sampling, and study design [16,17,18]. The aim of this study was to aggregate all publicly available datasets related to FSGS-associated glomerulus data and conduct a meta-analysis to establish a consensus list DEGs in this condition. This approach aimed to illuminate the underlying pathological pathways involved in FSGS. Briefly, array expression profiles underwent quality checks and normalization before being subjected to meta-analysis. The resulting list of DEGs (referred to as meta-DEGs) was further analyzed through methods such as functional enrichment analysis and the construction of a regulatory network. Additionally, a protein-protein interaction (PPI) network involving DEGs, their associated microRNAs (miRNAs), and transcription factors (TFs) was constructed and analyzed to identify potential molecules contributing to the pathogenesis of FSGS.
Materials and methods
Screening, quality checking, and selection of expression profiles
The NCBI Gene Expression Omnibus (GEO) was screened using keywords “FSGS” and “focal and segmental glomerulosclerosis” to identify and download expression profiles relevant to FSGS. Datasets deposited before June 20, 2021, were initially screened based on predefined inclusion and exclusion criteria for inclusion in this meta-analysis.
Inclusion criteria comprised: (a) Human sample datasets, (b) Glomerulus sample profiles, (c) Datasets with control samples, and (d) Array expression datasets. Exclusion criteria comprised: (a) Non-human or non-FSGS expression profiles, (b) Non-glomerular expression profiles, (c) Datasets lacking control samples, and (d) PCR or RNA-sequencing expression profiles.
Each dataset selected underwent quality evaluation using an unsupervised strategy, specifically principal component analysis (PCA). This approach ensured the datasets included in the meta-analysis were robust and suitable for further analysis.
Meta-analysis of the selected expression profiles
We normalized the datasets, integrated them, performed meta-analysis, and identified the list of meta-DEGs using Network Analyst, a web interface tailored for integrative meta-analysis [16]. Each dataset underwent normalization using quantile normalization and was analyzed individually. To address batch effects across datasets, the ComBat procedure was applied. Subsequently, Cochran’s Q-test was conducted on the merged dataset to assess statistical heterogeneity. Finally, a meta-analysis was performed using a random-effects model to consolidate findings across datasets and derive robust conclusions regarding the differential expression of genes associated with FSGS. This systematic approach ensured that the results were statistically rigorous and minimized biases introduced by dataset-specific variations and batch effects [19,20,21]. A false discovery rate (Benjamini–Hochberg method) cutoff of < 0.05 was applied for dataset analysis and identification of DEGs. To obtain the log2 fold change of the meta-DEGs in each dataset, the included datasets were also subjected to analysis with a false discovery rate cutoff of < 0.05, indicating substantial expression differences between experimental groups. The minimal change disease (MCD) and membranous nephropathy (MN) samples from the dataset GSE108109 were subjected to analysis and the DEGs were extracted considering a false discovery rate cutoff of < 0.05.
Meta-DEG’s enrichment analysis
The meta-DEG’s related gene ontology (GO) terms including molecular function, biological process, and cellular component terms, were obtained using the ClueGO plugin in Cytoscape [22, 23]. Likewise, the Reactome database was used for recognizing the meta-DEG’s related biological pathways. The terms were obtained from the most updated (July 2022) version of ontology/pathway databases. The significant (FDR < 0.05) terms were extracted and considered for further analysis. The ClueGO plugin in Cytoscape was utilized to construct a network comprising interconnected GO terms.
Regulatory network construction, analysis, and hub gene identification
To construct and analyze a regulatory network, initially, the list of meta-DEGs was uploaded to the STRING server to construct the protein-protein interaction network using a confidence cutoff of 0.4. Subsequently, meta-DEGs associated with microRNAs (miRNAs) and transcription factors (TFs) were identified using MiRTarBase (Release 7) [24] and TRRUST (Version 2) [25] databases. The regulatory network, comprising meta-DEGs, their related miRNAs, and TFs as their upstream regulatory elements, was visualized and analyzed using Cytoscape. Following this, the cytoHubba plugin [26] in Cytoscape was employed to identify top molecules in each category based on their degree of centrality.
Real-time quantitative polymerase chain reaction
In this step, several DEGs having a role in angiogenesis regulation including disintegrin and metalloproteinase with thrombospondin motif (ADAMTS1), platelet factor 4 (PF4), Early growth response factor 1 (EGR1), and epidermal growth factor (EGF) were selected for the gene expression analysis. The selection criteria were based on an extensive review of the literature, which confirmed the established roles of these genes in the regulation of angiogenesis. Additionally, the combined effect size from the meta-analysis and the log fold change values of the DEGs were also considered in the selection process.
Briefly, RNA was extracted from paraffin-embedded renal biopsies of 24 FSGS patients and 15 unaffected parts of tumor nephrectomy samples using an RNA extraction kit (Azmaelixir, Tehran, Iran) following the manufacturer’s instructions. The concentration of extracted RNA was measured using a nanodrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). Subsequently, cDNA synthesis was performed using a first-strand cDNA synthesis kit (YektaTajhiz, Tehran, Iran). Specific primers for the four selected DEGs were designed using AlleleID software (version 6.2); their sequences are provided in Table 1. RT-qPCR was conducted on an AB StepOnePlus System (ThermoFisher Scientific) with StepOnePlus software version 2.3. The reaction mixture included specific primers, cDNA template, water, and Real Q Plus 2x Master Mix Green with high ROX™ (Ampliqon, Odense, Denmark). The human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene served as an internal control for normalization. Relative expression levels were analyzed using the 2 − ΔΔCt method, and GraphPad Prism software (version 8.0.2) was utilized for data analysis. Statistical analysis employed an unpaired nonparametric t-test, and all data were presented as mean ± standard error of the mean (SEM), with significance set at P ≤ 0.05.
Results
Dataset screening, selection, and quality control: seven expression profiles were selected for the meta-analysis
After searching the specified keywords in the GEO dataset and applying filters (selecting expression profiling by array and Homo sapiens), we identified 18 microarray expression profiles. Among these, 8 profiles were derived from non-glomerular samples (tubule or whole blood), 3 profiles lacked healthy controls, and 7 profiles pertained to glomerular samples comprising 79 FSGS and 64 healthy kidneys, which were selected for further analysis. The GEO accessions of the selected expression profiles were GSE47183 (GPL14663), GSE47183 (GPL11670), GSE99340, GSE108109, GSE121233, GSE129973, and GSE104948. Detailed information on the selected datasets is provided in Table 2. Flow diagrams illustrating the study’s methodology and the process of microarray data identification, screening, and selection are presented in Figs. 1 and 2.

Flow diagram representing different steps of the present meta-analysis

Flow diagram of data selection. Different steps of microarray dataset selection including identification, screening, and eligibility are shown in this flow diagram
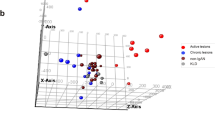
PCA, a widely used data reduction method, was employed to assess dataset quality and identify potential outliers among the samples [27]. Following PCA execution, outlier removal, normalization, and batch effect correction, a total of 79 FSGS and 64 control samples were included in the meta-analysis. PCA plots for each dataset are depicted in Figure S1 (supplementary figures file), with the final PCA plot illustrating the results of batch effect removal shown in Fig. 3A. Additionally, a three-dimensional PCA plot was constructed to confirm the differentiation between case and control samples following batch effect correction (Figure S2).

Data preprocessing and meta-analysis; (A) PCA plots before and after batch effect removal showing the differences and similarities between the samples and density plots against log2 of read counts, before and after batch effect removal depicting the relative distribution of different counts in each group. (B) The UpSet diagram representing the intersection of DEGs in datasets. (C) The heatmap of top 50 DEGs according to the adjusted p-value
DEG identification and meta-analysis: 2898 DEGs were identified as FSGS meta-DEGs
The meta-analysis was successfully conducted using the random effects model, selected based on the Cochran’s Q-test plot results [21, 28]. The outcome of the meta-analysis yielded a list of 2898 differentially expressed genes (DEGs), referred to as meta-DEGs, comprising 1584 upregulated and 1314 downregulated genes. Detailed results of the meta-analysis, including the list of meta-DEGs and their effect sizes, can be found in Table S2 of supplementary tables file. The UpSet diagram, illustrating the intersection of DEGs across datasets, and the heatmap displaying the top 50 meta-DEGs based on adjusted p-values, are presented in Fig. 3B and C, respectively.
Functional enrichment analysis: carboxylic acid catabolic process, homeostasis, extracellular matrix organization, and regulation of angiogenesis were some of the top terms
Figure 4 depicts the enrichment map showing significant biological process terms enriched for the FSGS meta-DEGs. These meta-DEGs were notably enriched in several biological processes including carboxylic acid catabolic process, phosphate-containing compound metabolic process, positive and negative regulation of vascularization, homeostatic process, apoptotic process, mitotic cell cycle, extracellular matrix organization, platelet signaling, and TCA cycle. The results from Reactome pathway enrichment also corroborated these findings, highlighting terms related to extracellular matrix organization, platelet degranulation, cell cycle phase transition, homeostasis, and metabolism. In terms of molecular functions, the FSGS meta-DEGs were enriched in functions such as transition metal ion binding, protein kinase binding, NAD binding, and RNA binding. Furthermore, based on cellular component gene ontology analysis, these genes predominantly function within intracellular organelle lumen, secretory granule lumen, collagen-containing extracellular matrix, mitochondrial matrix, focal adhesion, and nucleus. Table 3 lists the top 10 enriched biological process terms and pathways identified in this analysis.

The network of connected significant enriched biological process terms for the FSGS meta-DEGs. Each node represents a GO biological process, colored to indicate GO groups. Edges between nodes signify relationships based on the similarity of associated genes
Co-regulatory network construction and hub molecule identification: top DEGs, and their related regulatory elements were spotted in the co-regulatory network
From a systems biology perspective, alongside the PPI network of meta-DEGs, a co-regulatory network was constructed by incorporating two additional regulatory elements: microRNAs (miRNAs) and transcription factors (TFs). The constructed network is accessible on the Network Data Exchange (NDEx) server via the web address: [https://www.ndexbio.org/#/network/56205b7f-d500-11ec-b397-0ac135e8bacf?accesskey=8d34e9d83965d1f1d377f803931bca925ed678e40614b813f3ec7720c48e371b]. This comprehensive network comprises 5921 nodes and 186,745 edges, encompassing 2859 DEGs, 2688 miRNAs, and 374 TFs, with degrees ranging from 1 to 1511. Within each group, hub DEGs were selected based on their degree centrality scores (see Table 4). Among miRNAs, the top 5 regulators targeting FSGS DEGs were identified as hsa-mir-1-3p, hsa-mir-16-5p, hsa-mir-124-3p, hsa-mir-155-5p, and hsa-mir-34a-5p. Similarly, the top transcription factors influencing the expression of FSGS DEGs included specificity protein 1 (SP1), transcription factor p65 (RELA), E2F transcription Factor 1 (E2F1), specificity protein 3 (SP3), and MYC Proto-Oncogene (MYC). This integrated approach provides insights into the complex regulatory interactions underlying FSGS pathophysiology, facilitating a deeper understanding of the molecular mechanisms involved. In addition, a constructed PPI network including only the FSGS meta-DEGs, presenting the hub DEGs (based on degree centrality) and the DEGs involved in the angiogenesis regulatory pathways is shown in Fig. 5A.

(A) The PPI network of FSGS meta-DEGs. The square includes all the DEGs involved in the regulation of angiogenesis. The DEGs in the small rectangle are the top 20 DEGs based on degree centrality in the network. Interestingly, most of the top DEGs (based on degree centrality) are involved in angiogenesis regulation. The labeled (star) DEGs were selected for expressional analysis in the FSGS tissue samples. (B) The expression analysis results of 4 involved DEGs in the angiogenesis. ADAMTS1 and EGR1 showed a significant dysregulation in the FSGS samples
Real-time quantitative polymerase chain reaction: ADAMTS1 and EGFR showed a significant dysregulation in the FSGS samples
We conducted real-time quantitative polymerase chain reaction (RT-qPCR) to validate the results of our in-silico analysis. Specifically, we selected several candidate DEGs involved in either promoting or inhibiting angiogenesis to assess their expression in FSGS samples. According to our in-silico analysis, angiogenesis inhibitors such as ADAMTS1 and PF4 exhibited an up-regulated pattern in FSGS patients. Conversely, angiogenesis promoters like EGR1 and EGF were down-regulated in FSGS samples (Fig. 5A). To validate these findings, we evaluated the expression levels of these four genes in FSGS and normal tissue samples. Detailed demographic and clinical characteristics of the patients are provided in Table S3 of supplementary tables file. As depicted in Fig. 5B, the RT-qPCR results corroborated our in-silico analysis. Specifically, mRNA expression levels of ADAMTS1 and EGR1 were significantly higher and lower, respectively, in FSGS samples compared to the control group [ADAMTS1: 3.872 ± 0.9723 vs. 1.319 ± 0.4129, P = 0.0334; EGR1: 0.837 ± 0.2610 vs. 1.342 ± 0.3077, P = 0.0297]. However, no significant differences were observed in the expression of EGF and PF4 between FSGS and normal groups [EGF: 2.406 ± 0.6372 vs. 1.334 ± 0.5565, P = 0.5711; PF4: 1.195 ± 0.3321 vs. 1.521 ± 0.4624, P = 0.4395]. These findings validate the differential expression patterns of selected DEGs related to angiogenesis in FSGS, as identified through both computational and experimental approaches.
Discussion
Despite extensive research on FSGS, understanding the molecular mechanisms of its pathogenesis remains a critical area for exploration. Systems biology offers valuable tools to unravel these mechanisms and identify novel therapeutic targets. In this context, meta-analysis of transcriptomic datasets represents a powerful approach to gain insights into disease progression. Integrating expression data from multiple independent studies enhances statistical power and promotes more robust findings. In our study, we conducted a meta-analysis of publicly available FSGS microarray datasets that included human glomerulus samples. Following an exhaustive search and quality control assessment using PCA [29], meta-analysis was performed on seven FSGS datasets using the random effects model. The resulting list of DEGs represents a consensus set associated with the disease and underwent further analysis. Subsequently, we obtained PPI network data from the STRING server and identified DEG-associated TFs and miRNAs. These elements were integrated to construct a regulatory network. The primary goal of incorporating these regulatory elements alongside proteins is to elucidate co-regulatory mechanisms and develop a comprehensive interactome model specific to FSGS. This integrated approach aims to provide a deeper understanding of the molecular networks driving FSGS pathogenesis, potentially uncovering new avenues for therapeutic intervention [8, 30].
By assessing the degree of centrality in the constructed network, several DEGs, miRNAs, and TFs were identified as hub molecules, suggesting their potential roles in the underlying pathological pathways. One such hub molecule identified was fibronectin 1 (FN1), a glycoprotein found in the extracellular matrix and plasma. FN1 has been extensively implicated in various kidney diseases, including diabetic nephropathy, glomerular disease with fibronectin deposition (GFND), and renal fibrosis [31,32,33,34]. These conditions often involve pathological processes where fibronectin plays a crucial role in tissue remodeling and fibrosis progression. Although effective therapies targeting fibrosis progression remain limited, some studies have shown promising results. For instance, inhibition of fibronectin polymerization using a small peptide called pUR4 has been demonstrated to attenuate fibrosis in injured kidneys [35]. This approach highlights the potential therapeutic significance of targeting FN1 and its interactions in mitigating fibrotic processes in kidney diseases like FSGS. STAT3, identified as another hub molecule in the constructed FSGS regulatory network, is a pivotal component of the JAK-STAT signaling pathway known for its heightened transcriptional activity in autoimmune disorders and cancers [36]. Recent studies have increasingly highlighted the role of STAT3 in the progression of various kidney diseases. Research indicates that genetic knockout or knockdown of STAT3 in kidney disease models may confer beneficial effects [37,38,39]. For example, experiments have shown that inhibiting JAK2 or STAT3 activation in animal models can mitigate the FSGS-induced increase in albumin permeability [40]. These findings underscore STAT3 as a potential therapeutic target in FSGS and suggest that targeting the JAK-STAT pathway could potentially attenuate disease progression by modulating key signaling pathways involved in renal pathology.
In addition to the identified hub DEGs, several TFs and miRNAs were recognized as central molecules regulating the expression of DEGs within the FSGS network. These regulatory molecules represent potential targets for therapeutic intervention in FSGS. For instance, miR-155-5p was identified as a hub molecule targeting over 1000 DEGs in the FSGS network. Inhibition of miR-155-5p has been shown to attenuate kidney injury and impede the progression of renal fibrosis in animal models of FSGS [41]. This highlights miR-155-5p as a promising therapeutic target in FSGS, potentially offering a strategy to modulate disease progression by targeting its regulatory effects on multiple genes. Recent reviews, such as Zhao et al., discuss the emerging role of miRNAs in the clinical progression of chronic kidney diseases, underscoring their potential as diagnostic biomarkers and therapeutic targets [42]. This research suggests that targeting miRNAs like miR-155-5p could provide new avenues for treating FSGS and other kidney disorders characterized by dysregulated gene expression networks.
Based on the GO and Reactome pathway enrichment analyses, the meta-DEGs identified in our study were implicated in various biological pathways, particularly those related to the cell cycle, apoptosis, metabolism, wound healing, and angiogenesis. Specifically, terms such as vascularization, tube development, and positive and negative regulation of angiogenesis were prominently enriched (Fig. 4 and S5). These enrichment results indicate an imbalance between pro-angiogenic and anti-angiogenic factors in FSGS kidneys. Such dysregulation in the mechanisms governing vascular injury, repair, and homeostasis is well-recognized in kidney disorders [43]. According to previous experiments, renal microvascular disease, which plays a crucial role in the progression of renal diseases, becomes increasingly severe with the advancement of the illness [44, 45]. Furthermore, our decision to emphasize angiogenesis in our study was influenced by previous research demonstrating its involvement in the pathophysiology of diabetic kidney disease (DKD), which shares similarities with FSGS. Studies on DKD have shown an imbalance in the expression of angiogenic genes, highlighting the importance of vascular factors in kidney diseases such as FSGS [46]. Similarly, insufficient angiogenesis leading to hypoxia is recognized as a primary contributor to fibrosis in diseased kidneys. Maintaining a balance between proangiogenic and antiangiogenic factors is crucial for the integrity of the renal vascular network. Restoring normal angiogenic processes represents a promising strategy to address both hypoxia and fibrosis in kidney diseases [47]. Apart from the discussed kidney disease, there are two processes in which an imbalance between proangiogenic and antiangiogenic factors may lead to a pattern of FSGS including preeclampsia [48, 49] and glomerular disease after anti-VEGF use [50,51,52]. Additionally, angiogenesis plays a crucial role in numerous physiological and pathological processes, including tissue repair and inflammation. In the context of FSGS, aberrant angiogenesis may contribute to the pathogenesis of the disease by influencing renal vascular remodeling, inflammation, and fibrosis. Targeting these processes could represent a promising therapeutic strategy for various chronic kidney diseases, including FSGS.
Of note, to assess whether the dysregulation of angiogenesis-related genes is specific to FSGS glomeruli, we analyzed the GSE108109 dataset, including MCD and MN samples. The DEGs were examined through enrichment analysis, which showed no significant similarity to FSGS-associated terms. Additionally, a Venn diagram comparison of angiogenesis-related DEGs from FSGS versus MCD and MN DEGs indicated that only one-third and half of the angiogenesis-related DEGs were also dysregulated in MCD and MN, respectively (Tables S4-S7 & Figure S4 in the supplementary files). This observation might indicate the specificity of angiogenesis dysregulation in FSGS but not in other similar conditions like MCD and MN.
ADAMTS1 (a disintegrin and metalloproteinase with thrombospondin motif), for instance, exhibited up-regulation in FSGS samples (Log2FC ~ 1.5-2), as noted in previous studies [53, 54]. This VEGF inhibitor’s increased expression in patient kidneys suggests a suppression of angiogenesis in FSGS. Similarly, other anti-angiogenic factors such as platelet factor 4 (PF4) and proline-rich homeodomain protein (HHEX, another VEGFA regulator) were up-regulated, while several angiogenesis inducers including early growth response factor 1 (EGR1), epidermal growth factor (EGF), and plasminogen (PLG) were down-regulated in FSGS samples [55,56,57,58,59]. A comprehensive list of angiogenesis-related FSGS-DEGs, along with their effect size values, is provided in table S2 of supplementary tables file. Interestingly, the expression profile of the vascular endothelial growth factor (VEGF) family, well-known for its angiogenic properties, showed only a slight increase in FSGS kidneys (Log2FC ~ 0.2–0.9). This disturbance in angiogenesis warrants further investigation as a potential target to impede the progression of FSGS.
In the expression evaluation experiment, we selected four of the aforementioned DEGs and examined their expression in paraffin-embedded FSGS samples. As anticipated, ADAMTS1 was significantly up-regulated and EGR1 was significantly down-regulated in the FSGS samples. The other two DEGs, PF4 and PLG, also showed expected expression patterns in the FSGS samples, but the results were not statistically significant. One possible reason for this outcome could be the unavailability of fresh FSGS samples, as our experiment relied on paraffin-embedded tissues.
In terms of clinical translation of our findings, we hypothesize that restoring the balance of angiogenesis by targeting either the upregulated angiogenesis inhibitors (e.g., PF4, ADAMTS1) or the downregulated angiogenesis inducers (e.g., EGF, EGR1) has the potential to alleviate FSGS progression and restore a balanced angiogenic response in the kidney. Enhancing this balance could hold promise for mitigating FSGS progression through various mechanisms. Balanced blood vessel formation would improve nutrient delivery to kidney tissues, facilitating repair and regeneration. This approach could potentially offer new therapeutic avenues for treating FSGS and related kidney disorders [60]. Moreover, enhancing blood vessel density could alleviate the issue of hypoxia in FSGS kidneys, thereby promoting tissue healing [61]. Additionally, considering that dysregulated angiogenesis is implicated in chronic inflammation, a hallmark of FSGS, restoring a balanced angiogenic response may help regulate inflammatory processes. This could potentially reduce tissue damage and inflammation in the kidney, offering therapeutic benefits for managing FSGS and its associated complications [62].
In light of our findings, we hypothesize that dysregulation of angiogenesis pathways may play a role in the progression of FSGS. However, it remains unclear whether promoting or inhibiting angiogenesis would be therapeutically beneficial. Further studies are needed to define the precise impact of modulating angiogenesis on FSGS progression. This hypothesis is exploratory, as there is currently no literature directly supporting the therapeutic targeting of angiogenesis in FSGS. Future research will be crucial to determine whether interventions aimed at either promoting or reducing angiogenesis can effectively mitigate the disease.
Our study has several limitations that should be acknowledged. Firstly, we used qPCR exclusively to validate gene expression changes, which limited our exploration to mRNA levels without assessing protein expression, thus restricting our insight into the molecular mechanisms underlying FSGS. Future investigations should incorporate techniques such as immunohistochemistry to provide a more comprehensive analysis including protein levels.
Furthermore, our analysis relied only on microarray data and did not include RNA sequencing, potentially reducing the depth and resolution of our gene expression profiling. We know that microarrays offer various advantages, including the expression of thousands of genes simultaneously, low sample consumption, easy sample preparation, and control of experimental conditions. However, this technology has some disadvantages. These include competence required for data normalization and analysis, limited dynamic range, low sensitivity and competitive hybridization. Compared to microarrays, NGS offers greater flexibility, high sensitivity and dynamic range, no hybridization, and higher cost-effectiveness. Our decision to meta-analyses the available microarray data was based on the results of a systematic search of the GEO dataset. Therefore, if NGS datasets were available, we would certainly have used them and added them to our meta-analysis.
In terms of the source of the analyzed samples, GSE47183, GSE99340, GSE104948 and GSE108109 datasets were provided from the multicentre European Renal cDNA Bank-Kroener-Fresenius biopsy bank (ERCB) and GSE121233 and GSE129973 Samples were generated from Renal Biobank of National Clinical Research Center of Kidney Diseases at Jinling Hospital in China. Our FSGS samples were prepared from Iranian samples of Al-Zahra Hospital, Isfahan. Therefore, variability in RNA sources especially in the race of samples poses another limitation, which could impact the comparability of RNA changes across different platforms. In addition, the type of FSGS (primary, secondary or genetic types) was not available in the dataset information or their articles and in our samples, and this situation could introduce heterogeneity in the samples in the dataset pooling and their validation.
In order to improve future studies, in addition to trying to overcome the above limitations, it is necessary to include a wider range of glomerulopathies and facilitate a more comprehensive understanding of molecular mechanisms in different renal conditions.
Data availability
The analyzed datasets by the present study are available in the GEO repository, [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104948], [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129973], [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE47183], [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108109], [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99340],
[https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121233].
References
Sun K, Xie Q, Hao C-M. Mechanisms of scarring in focal segmental glomerulosclerosis. Kidney Dis. 2021;7(5):350–8.
Valencia VC, de La Cruz CO, Fuentes JB, Ramírez FF, Michel RP, Aragaki Y, Magaña IM, Villaseñor HP, Pérez MV, Cárdenas MG. Epidemiology of glomerular disease in adults: a database review. Gac Med Mex. 2014;150(5):403–8.
Awazu M, Yamada M, Asada N, Hashiguchi A, Kosaki K, Matsumura K. A girl with a mutation of the ciliary gene CC2D2A presenting with FSGS and nephronophthisis. CEN case Rep. 2022;11(1):116–9.
Kim JS, Han BG, Choi SO, Cha S-K. Secondary focal segmental glomerulosclerosis: from podocyte injury to glomerulosclerosis. BioMed research international 2016, 2016.
Shabaka A, Ribera AT, Fernández-Juárez G. Focal segmental glomerulosclerosis: state-of-the-art and clinical perspective. Nephron. 2020;144(9):413–27.
Sim JJ, Batech M, Hever A, Harrison TN, Avelar T, Kanter MH, Jacobsen SJ. Distribution of biopsy-proven presumed primary glomerulonephropathies in 2000–2011 among a racially and ethnically diverse US population. Am J Kidney Dis. 2016;68(4):533–44.
Komers R, Diva U, Inrig JK, Loewen A, Trachtman H, Rote WE. Study design of the phase 3 sparsentan versus irbesartan (DUPLEX) study in patients with focal segmental glomerulosclerosis. Kidney Int Rep. 2020;5(4):494–502.
Gholaminejad A, Fathalipour M, Roointan A. Comprehensive analysis of diabetic nephropathy expression profile based on weighted gene co-expression network analysis algorithm. BMC Nephrol. 2021;22(1):1–13.
Gholaminejad A, Ghaeidamini M, Simal-Gandara J, Roointan A. An integrative in silico study to Discover Key drivers in pathogenicity of focal and segmental glomerulosclerosis. Kidney Blood Press Res 2022:1–13.
Gholaminejad A, Gheisari Y, Jalali S, Roointan A. Comprehensive analysis of IgA nephropathy expression profiles: identification of potential biomarkers and therapeutic agents. BMC Nephrol. 2021;22(1):1–10.
Gholaminejad A, Roointan A, Gheisari Y. Transmembrane signaling molecules play a key role in the pathogenesis of IgA nephropathy: a weighted gene co-expression network analysis study. BMC Immunol. 2021;22(1):1–17.
Nadon R, Shoemaker J. Statistical issues with microarrays: processing and analysis. Trends Genet. 2002;18(5):265–71.
Fulcher BD, Arnatkeviciute A, Fornito A. Overcoming false-positive gene-category enrichment in the analysis of spatially resolved transcriptomic brain atlas data. Nat Commun. 2021;12(1):2669.
Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, Hudelle R, Qaiser T, Matson KJ, Barraud Q. Confronting false discoveries in single-cell differential expression. Nat Commun. 2021;12(1):5692.
Bero L, Rennie D. The Cochrane collaboration: preparing, maintaining, and disseminating systematic reviews of the effects of health care. JAMA. 1995;274(24):1935–8.
Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47(W1):W234–41.
Rung J, Brazma A. Reuse of public genome-wide gene expression data. Nat Rev Genet. 2013;14(2):89–99.
Haidich A-B. Meta-analysis in medical research. Hippokratia. 2010;14(Suppl 1):29.
Zhang Y, Jenkins DF, Manimaran S, Johnson WE. Alternative empirical Bayes models for adjusting for batch effects in genomic studies. BMC Bioinformatics. 2018;19(1):262.
Choi JK, Yu U, Kim S, Yoo OJ. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19(Suppl 1):i84–90.
Jha PK, Vijay A, Sahu A, Ashraf MZ. Comprehensive Gene expression meta-analysis and integrated bioinformatic approaches reveal shared signatures between thrombosis and myeloproliferative disorders. Sci Rep. 2016;6:37099.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W-H, Pagès F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3.
Chou C-H, Shrestha S, Yang C-D, Chang N-W, Lin Y-L, Liao K-W, Huang W-C, Sun T-H, Tu S-J, Lee W-H. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46(D1):D296–302.
Han H, Cho J-W, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46(D1):D380–6.
Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11–11.
Yeung KY, Ruzzo WL. Principal component analysis for clustering gene expression data. Bioinformatics. 2001;17(9):763–74.
Zhou G, Stevenson MM, Geary TG, Xia J. Comprehensive Transcriptome Meta-analysis to characterize host Immune responses in Helminth infections. PLoS Negl Trop Dis. 2016;10(4):e0004624.
Abedi M, Fatehi R, Moradzadeh K, Gheisari Y. Big data to knowledge: common pitfalls in transcriptomics data analysis and representation. RNA Biol. 2019;16(11):1531–3.
Hojjati F, Roointan A, Gholaminejad A, Eshraghi Y, Gheisari Y. Identification of key genes and biological regulatory mechanisms in diabetic nephropathy: Meta-analysis of gene expression datasets. Nefrología 2022.
Livingston MJ, Ding H-F, Huang S, Hill JA, Yin X-M, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy. 2016;12(6):976–98.
Alvarez ML, DiStefano JK. Functional characterization of the plasmacytoma variant translocation 1 gene (PVT1) in diabetic nephropathy. PLoS ONE. 2011;6(4):e18671.
Kliewe F, Kaling S, Lötzsch H, Artelt N, Schindler M, Rogge H, Schröder S, Scharf C, Amann K, Daniel C. Fibronectin is up-regulated in podocytes by mechanical stress. FASEB J. 2019;33(12):14450–60.
Castelletti F, Donadelli R, Banterla F, Hildebrandt F, Zipfel PF, Bresin E, Otto E, Skerka C, Renieri A, Todeschini M. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proceedings of the National Academy of Sciences 2008, 105(7):2538–2543.
Bowers SL, Davis-Rodriguez S, Thomas ZM, Rudomanova V, Bacon WC, Beiersdorfer A, Ma Q, Devarajan P, Blaxall BC. Inhibition of fibronectin polymerization alleviates kidney injury due to ischemia-reperfusion. Am J Physiology-Renal Physiol. 2019;316(6):F1293–8.
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19(1):1–19.
Pace J, Paladugu P, Das B, He JC, Mallipattu SK. Targeting STAT3 signaling in kidney disease. Am J Physiology-Renal Physiol. 2019;316(6):F1151–61.
Sun H-J, Xiong S-P, Cao X, Cao L, Zhu M-Y, Wu Z-Y, Bian J-S. Polysulfide-mediated sulfhydration of SIRT1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 NF-κB and STAT3. Redox Biol. 2021;38:101813.
Lu T-C, Wang Z-H, Feng X, Chuang PY, Fang W, Shen Y, Levy DE, Xiong H, Chen N, He JC. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int. 2009;76(1):63–71.
Sharma M, Zhou J, Gauchat J-F, Sharma R, McCarthy ET, Srivastava T, Savin VJ. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Translational Res. 2015;166(4):384–98.
Liu G, He L, Yang X, Tang L, Shi W, She J, Wei J. MicroRNA-155-5p aggravates Adriamycin-Induced Focal Segmental Glomerulosclerosis through Targeting Nrf2. Nephron 2022:1–12.
Zhao H, Ma S-X, Shang Y-Q, Zhang H-Q, Su W. microRNAs in chronic kidney disease. Clin Chim Acta. 2019;491:59–65.
Tanabe K, Wada J, Sato Y. Targeting angiogenesis and lymphangiogenesis in kidney disease. Nat Rev Nephrol. 2020;16(5):289–303.
Futrakul N, Butthep P, Futrakul P. Altered vascular homeostasis in chronic kidney disease. Clin Hemorheol Microcirc. 2008;38(3):201–7.
Futrakul N, Butthep P, Laohareungpanya N, Chaisuriya P, Ratanabanangkoon K. A defective angiogenesis in chronic kidney disease. Ren Fail. 2008;30(2):215–7.
Fadini GP, Albiero M, Bonora BM, Avogaro A. Angiogenic abnormalities in diabetes mellitus: mechanistic and clinical aspects. J Clin Endocrinol Metabolism. 2019;104(11):5431–44.
Kawakami T, Mimura I, Shoji K, Tanaka T, Nangaku M. Hypoxia and fibrosis in chronic kidney disease: crossing at pericytes. Kidney Int Supplements. 2014;4(1):107–12.
Kwiatkowska E, Stefańska K, Zieliński M, Sakowska J, Jankowiak M, Trzonkowski P, Marek-Trzonkowska N, Kwiatkowski S. Podocytes-the most vulnerable renal cells in Preeclampsia. Int J Mol Sci 2020, 21(14).
Fritzsche FR, Ramach C, Soldini D, Caduff R, Tinguely M, Cassoly E, Moch H, Stewart A. Occupational health risks of pathologists–results from a nationwide online questionnaire in Switzerland. BMC Public Health. 2012;12:1054.
Phadke G, Hanna RM, Ferrey A, Torres EA, Singla A, Kaushal A, Kalantar-Zadeh K, Kurtz I, Jhaveri KD. Review of intravitreal VEGF inhibitor toxicity and report of collapsing FSGS with TMA in a patient with age-related macular degeneration. Clin Kidney J. 2021;14(10):2158–65.
Fleming K, McGuinness J, Kipgen D, Glen H, Spiliopoulou P. A Case of Lenvatinib-Induced Focal Segmental Glomerulosclerosis (FSGS) in Metastatic Medullary Thyroid Cancer. Case Rep Oncol Med 2018, 2018:6927639.
Furuto Y, Hashimoto H, Namikawa A, Outi H, Takahashi H, Horiuti H, Honda K, Shibuya Y. Focal segmental glomerulosclerosis lesion associated with inhibition of tyrosine kinases by lenvatinib: a case report. BMC Nephrol. 2018;19(1):273.
Xu Z, Yu Y, Duh EJ. Vascular endothelial growth factor upregulates expression of ADAMTS1 in endothelial cells through protein kinase C signaling. Investig Ophthalmol Vis Sci. 2006;47(9):4059–66.
Lambert J, Makin K, Akbareian S, Johnson R, Alghamdi AA, Robinson SD, Edwards DR. ADAMTS-1 and syndecan-4 intersect in the regulation of cell migration and angiogenesis. J Cell Sci. 2020;133(7):jcs235762.
Seminars in thrombosis and hemostasis: 2004. Copyright© 2004 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New ….
Kershaw RM, Roberts D, Wragg J, Shaaban AM, Humphreys E, Halsall J, Price L, Bicknell R, Gaston K, Jayaraman P-S. Proline-Rich Homeodomain protein (PRH/HHEX) is a suppressor of breast tumour growth. Oncogenesis. 2017;6(6):e346–346.
Wang B, Guo H, Yu H, Chen Y, Xu H, Zhao G. The role of the transcription factor EGR1 in cancer. Front Oncol. 2021;11:642547.
Möller B, Rasmussen C, Lindblom B, Olovsson M. Localized expression of the angiogenic growth factors vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF-2) and epidermal growth factor (EGF) and their receptors in normal cycling human endometrium. Mol Hum Reprod. 2001;7(1):65–72.
Fang J, Chopp M, Xin H, Zhang L, Wang F, Golembieski W, Zhang ZG, He L, Liu Z. Plasminogen deficiency causes reduced angiogenesis and behavioral recovery after stroke in mice. J Cereb Blood Flow Metabolism. 2021;41(10):2583–92.
Lerman LO, Chade AR. Angiogenesis in the kidney: a new therapeutic target? Curr Opin Nephrol Hypertens. 2009;18(2):160–5.
Tanaka S, Tanaka T, Nangaku M. Hypoxia and dysregulated angiogenesis in kidney disease. Kidney Dis. 2015;1(1):80–9.
De Vriese AS, Wetzels JF, Glassock RJ, Sethi S, Fervenza FC. Therapeutic trials in adult FSGS: lessons learned and the road forward. Nat Rev Nephrol. 2021;17(9):619–30.
Ju W, Greene CS, Eichinger F, Nair V, Hodgin JB, Bitzer M, Lee Y-s, Zhu Q, Kehata M, Li M. Defining cell-type specificity at the transcriptional level in human disease. Genome Res. 2013;23(11):1862–73.
Shved N, Warsow G, Eichinger F, Hoogewijs D, Brandt S, Wild P, Kretzler M, Cohen CD, Lindenmeyer MT. Transcriptome-based network analysis reveals renal cell type-specific dysregulation of hypoxia-associated transcripts. Sci Rep. 2017;7(1):1–17.
Grayson PC, Eddy S, Taroni JN, Lightfoot YL, Mariani L, Parikh H, Lindenmeyer MT, Ju W, Greene CS, Godfrey B. Metabolic pathways and immunometabolism in rare kidney diseases. Ann Rheum Dis. 2018;77(8):1226–33.
Hu S, Han R, Shi J, Zhu X, Qin W, Zeng C, Bao H, Liu Z. The long noncoding RNA LOC105374325 causes podocyte injury in individuals with focal segmental glomerulosclerosis. J Biol Chem. 2018;293(52):20227–39.
GSE129973: Gene Expression Omnibus (GEO) dataset. National Center for Biotechnology Information (NCBI). Available from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129973
Acknowledgements
We thank members of the Regenerative Medicine Research Center for their help in some parts of the bioinformatic analysis steps.
Funding
This work was supported by the Isfahan University of Medical Sciences [grant number 140013].
Author information
Authors and Affiliations
Contributions
A.R. participated in the design, screening, selection, meta-analysis, interpretation of data, and drafting of the manuscript. A.G. participated in the study design, performing the meta-analysis, data interpretation, and drafting of the manuscript. M.G. performed the real-time PCR experiment and participated in drafting the manuscript. P.Y. contributed to the data collection, data analysis, and drafting of the manuscript. A.N. contributed to the interpretation of the analyzed data, providing the FSGS samples and preparing the manuscript. Y.G. participated in this work by analyzing the obtained results and preparing the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in this study were in accordance with the ethical standards of the institutional and national research committee [IR.MUI.MED.REC.1400.063].
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Roointan, A., Ghaeidamini, M., Yavari, P. et al. Transcriptome meta-analysis and validation to discovery of hub genes and pathways in focal and segmental glomerulosclerosis. BMC Nephrol 25, 293 (2024). https://doi.org/10.1186/s12882-024-03734-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-024-03734-4