
Abstract
Fibronectin (FN) glomerulopathy (FNG), a rare autosomal hereditary renal disease, is characterized by proteinuria resulting from the massive accumulation of FN in the glomeruli. It typically affects individuals aged 10–50 years. In this report, we describe the case of a 57-year-old man who was diagnosed with FNG through genetic analysis and histological examination that revealed membranoproliferative glomerulonephritis. Despite treatment with prednisolone, the therapeutic response was unsatisfactory. Prednisolone was subsequently tapered and discontinued because the patient had pulmonary thromboembolism. Subsequent comprehensive genetic testing, which was initially not conducted because the patient’s parents did not have a history of kidney disease, identified a known disease-causing variant in the FN1 gene, indicating a de novo variant. FNG was further confirmed by positive staining of glomeruli with FN using an IST-4 antibody. Although corticosteroid therapy is commonly employed as the initial treatment for MPGN, its appropriateness depends on the underlying etiology. Thus, clinicians must be aware of potential rare genetic causes underlying MPGN.
Similar content being viewed by others


Background
Fibronectin (FN) glomerulopathy (FNG) is an uncommon autosomal dominant kidney disease. It is characterized by proteinuria, and some patients also exhibit nephrotic syndrome. Approximately half of the patients present with hematuria and hypertension, and 25% progress to end-stage renal disease within 15–20 years of disease onset, necessitating renal replacement therapy [1]. The precise number of cases is unknown, primarily due to the rarity of the disease, with no case reports or prevalence rates documented in previous reviews. In our investigation, we conducted a basic survey using PubMed searches. Specifically, a MeSH search was performed employing the keywords "glomerulopathy with fibronectin deposits" to encompass all available reports to date. We identified a total of approximately 113 cases, including those reported in case series. FNG is caused by variants in the FN1 gene located on chromosome 2. While some FNG cases have been reported to display membranoproliferative glomerulonephritis (MPGN)-like lesions [2], previous case reports lacked concurrent discussion of histology and genotype. Simultaneous consideration of these factors is essential for accumulating genotype–phenotype correlation data in similar cases in the future. Currently, no specific treatment is available for this disease; although previous reports [3] have mentioned the potential efficacy of steroids, their definitive role remains uncertain. In this report, we present a case of FNG with a pathological diagnosis of MPGN. The diagnosis was confirmed by retrospective genetic analysis after the initiation of steroid therapy because the patient’s parents did not have a history of kidney disease.
Case presentation
A 57-year-old Japanese man presented to our department because of urinary protein and renal dysfunction identified during a routine medical check-up. He had a history of hematuria since his 20 s. At the age of 53, significant urinary protein (2 +) became apparent; however, it had not been thoroughly investigated. The patient had mild hypertension, with a systolic blood pressure of approximately 130 mmHg. While his parents had no history of kidney disease, his maternal grandmother had kidney disease; however, specific details were unknown. In addition, a maternal cousin had undergone kidney transplantation in her 30 s. On the initial visit, laboratory examinations revealed mild renal dysfunction (serum creatinine [s-Cr] 1.03 mg/dL), proteinuria (urinary protein creatinine ratio [UPCR] 1.21 g/gCr), and hematuria (20–29 red blood cells [RBCs]/high-power field). Serum immunological examination revealed a weakly positive PR3-ANCA level of 4.9 U/L. Serum complement levels (C3, C4, and CH50) were within the normal range (Table 1). To confirm the diagnosis, a percutaneous renal biopsy was performed.
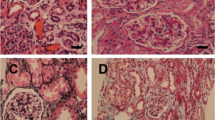
Figures 1, 2 and Supplementary Fig. 1 present the renal biopsy tissue. Light microscopy examination detected 30 glomeruli. Diffuse mesangial cell proliferation was observed, accompanied by thickening of the basement membrane, extensive mesangial infiltration, and doubling. Both Congo-red and Direct fast scarlet (DFS) staining, which are used to detect amyloid components, yielded negative results, and no staining bias was observed for either kappa or lambda. Immunofluorescent staining revealed mild glomerular deposition of IgG only (Supplementary Fig. 1). Furthermore, electron microscopy revealed electron-dense deposits (EDD) with high density in the paramesangial area and subendothelium, which appeared to consist of amorphous components, with less apparent fibrillary material (Fig. 2).

Light and electron microscopic examination. A and B Diffuse mesangial cell proliferation and increased mesangial matrix (HE and PAS staining, respectively, × 200). C PAM staining revealed thickening of the basement membrane with extensive mesangial infiltration and doubling. The lobulated segment exhibited scattered areas with methenamine silver-negative staining. (× 200). D MT staining showed a mild enhancement of trichrome red consistent with the lobulated region (× 200). E Congo-red staining yielded negative results. F Fibronectin (IST-4) staining exhibited a diffuse positive signal in the glomeruli, predominantly in the mesangial region (× 200). HE, hematoxylin and eosin staining; PAS, periodic acid-Schiff; PAM, periodic acid-methenamine-silver; MT, Masson trichrome staining

Electron microscopy (EM) of the kidney biopsy specimen. A High electron density deposits were found in the paramesangial area and subendothelium (EM × 1000). B Highly magnified image of the area encircled by the yellow square in image 2A
Following the identification of membranoproliferative pattern GN, the patient was diagnosed with idiopathic MPGN. He was readmitted to the hospital, and oral prednisolone treatment at a dose of 60 mg/day (1 mg/kg/day) was initiated. Upon admission, laboratory results indicated s-Cr level of 1.11 mg/dL, proteinuria of 0.78 g/gCr, and hematuria of 10–19 RBCs/HPF. During the hospitalization, the patient’s urinary protein levels decreased, and after 25 days, he was discharged with the prednisolone dosage reduced to 40 mg/day. However, his renal function gradually deteriorated during outpatient follow-up. During a visit to the outpatient clinic after discharge, his blood pressure dropped to 115/88 mmHg, whereas the urinary protein level remained at approximately 0.5 g/gCr. This may be partly caused by a pre-renal factor attributed to the use of diuretics (20 mg/day furosemide) to manage water retention associated with corticosteroid administration. Owing to the perceived limited therapeutic effect of prednisolone, the dose was gradually reduced to 20 mg/day. Furthermore, considering the COVID-19 pandemic at the time, there were also concerns regarding its effect.
On June 2, 2020, the patient presented to the emergency department with sudden right-sided abdominal pain. Laboratory tests revealed high D-dimer levels (67.83 μg/mL), and contrast-enhanced computed tomography (CT) confirmed the presence of thrombi in both the inferior pulmonary arteries and right inferior femoral vein. Pulmonary thromboembolism (PE) was established as a new event following the initiation of prednisolone therapy, as no evident thrombus formation was observed in a previous CT scan conducted on January 4. Following consultation with a cardiologist, apixaban therapy was initiated. Oxygen administration was gradually reduced over a few days, and the patient maintained a stable respiratory function without hypoxemia while breathing room air. During the treatment for pulmonary thromboembolism, the s-Cr levels rapidly increased to 1.3 mg/dL, and urinary protein transiently rose to 2.3 g/gCr. These changes can be partly attributed to the high blood pressure (increasing from a baseline of 120/80 mmHg to 140/100 mmHg) and high renal vein pressure caused by increased right ventricular system venous pressure. Considering that prednisolone lacked therapeutic efficacy and the presence of thromboembolism, prednisolone was further tapered and ultimately discontinued on January 4, 2021.
Given that the patient’s parents and first-degree relatives had no history of renal dysfunction or urinary abnormalities and the patient presented with MPGN without hypocomplementemia or any history suggestive of atypical hemolytic uremic syndrome (aHUS), genetic causes, including abnormalities in the complement system, was initially unlikely. Consequently, genetic testing was not performed at the outset. However, we subsequently conducted a comprehensive genetic screening using targeted next-generation sequencing (NGS) covering exons and splicing regions of 121 known genes responsible for major inherited kidney diseases (Supplementary Table) [4]. The results revealed a known in-frame deletion variant, c.4415_4417del: p.(Pro1472del), which had previously been reported as a disease-causing variant in FNG [5]. Immunostaining with FN (IST-4: Monoclonal anti-fibronectin antibody produced in mouse, clone IST-4, SIGMA-ALDRICH, F0916) demonstrated diffuse positive staining in the glomeruli, predominantly in the mesangial region, further confirming the diagnosis of FN nephropathy. The patient’s son, who had occasional episodes of microscopic haematuria during medical check-ups, also underwent genetic testing, and the same FN1 variant was identified. The co-segregation pattern provided additional support for the pathogenicity of the identified variant. After discontinuing the steroid therapy, the patient’s s-Cr level remained stable, ranging from approximately 1.3–1.4 mg/dL, and urinary protein levels were well controlled below 0.25 g/gCr with conservative treatment primarily using angiotensin II receptor blockers (ARBs). Notably, even without the use of anticoagulants, no signs of hypoxia were observed.
Discussion and conclusions
We encountered a case of FNG in a 57-year-old male patient who exhibited MPGN pathology, which was subsequently confirmed through genetic testing. The diagnosis was substantiated by both histological examination and genetic co-segregation analysis. Notably, the patient’s parents had no recorded history of renal disease or abnormal urinalysis, suggesting a probable de novo variant. Unfortunately, due to the parents’ demise, further verification was challenging.
FN is a high-molecular-weight glycoprotein component of the extracellular matrix. It exists as a circulating dimer and exhibits binding affinity toward heparin and integrins [6]. In normal physiological conditions, FN is primarily synthesized by mesangial cells in the kidney and liver. However, the precise mechanisms underlying the pathogenesis of FN accumulation remain incompletely understood. We hypothesized the involvement of the formation of FN mutants that are not excreted or the attachment of circulating factors. The deposits primarily consist of soluble plasma-derived FN rather than the insoluble cellular form. In vitro studies have demonstrated impaired binding of this FN variant to heparin on podocytes and endothelial cell surfaces [6]. Consequently, it exhibited the ability to induce endothelial cell expansion and podocyte cytoskeletal reorganization [5]. Another proposed mechanism involves impaired FN catabolism. Furthermore, the precipitate contains high levels of fibulin 1 and 5, which can bind to and/or regulate FN.
The typical pathological findings were as follows: periodic acid-Schiff (PAS)-positive deposits in the subendothelium and mesangium, hyperplasia of mesangial cells, and glomerulomegaly by light microscopy, negative or very light staining by fluorescent antibody, and unstructured granular deposits in the subendothelium and mesangium by electron microscopy, partially surrounded by bright bundles of fine fibers. In this specific case, we initially did not consider FN as a potential differential diagnosis. This was due to the presentation of membranoproliferative pattern GN with mild IgG deposition, along with the presence of amorphous EDD in the subendothelial or paramesangial region, which we initially considered as nonspecific. However, in retrospect, these findings support the diagnosis of FN, except there was no apparent family history.
FN plays a crucial role in cellular processes such as cell growth, differentiation, migration, and tissue remodeling, including wound healing in response to cellular injury [7]. Therapeutically, steroids acted on inflammatory cytokines and immune cells, making them effective in treating a broad spectrum of diseases. However, steroid therapy is associated with various adverse events, such as hypertension, diabetes, obesity, and increased susceptibility to infections [8]. In the present case, renal function deteriorated during steroid therapy. Although diuretic use may have contributed to this decline, renal function remained worse even after discontinuing both the diuretic and prednisolone therapy (Fig. 3). However, no definitive increase in proteinuria was noted, partly due to the beneficial effects of ARBs presumably. The transient decrease in urinary protein during 3 weeks of hospitalization was likely attributed to the effects of a low-salt diet and bed rest, rather than the direct effect of prednisolone therapy.

Clinical progression chart. Renal biopsy was performed during the initial hospitalization, followed by the initiation of prednisolone therapy at a dosage of 60 mg (1 mg/kg/day) on subsequent admission. However, renal function did not improved, resulting in a gradual reduction of the steroid dose
The p.(Pro1472del) variant identified in the present case was reported in two Japanese families [5], and the variant was located in the integrin-binding domain. In the literature, a member of family 4 developed end-stage kidney disease in their 40 s, whereas a member of family 10 presented with severe proteinuria, characterized by a UPCR level of approximately 5 g/gCr. By contrast, our case exhibited a relatively milder phenotype. These cases highlight the phenotypic variability associated with the variant and suggest the potential involvement of other modifier genes or environmental factors.
Corticosteroid therapy is commonly attempted in cases with a histological diagnosis of MPGN [9]. A study reported successful treatment in FNG cases with nephrotic-level proteinuria [10]. However, in the present case, we did not observe a clear treatment response, and only adverse events were evident. Steroids are known to enhance the production of coagulation factors and fibrinogen, consequently augmenting blood coagulation capacity and predisposing to thrombus formation [11]. This was considered as a potential side effect in this particular case. Currently, no specific treatment is available for FNG, and angiotensin-converting enzyme inhibitors and ARBs are generally used for renal protection. The initiation of steroid therapy for FNG should be carefully considered on an individual basis.
Although the in-frame deletion in FN1 identified in this study was previously reported, the accompanying pathological tissue findings were not presented [5]. Therefore, this report presents the first example of histological evidence corresponding to the p.(Pro1472del) mutation, which resulted in MPGN.
In conclusion, FNG must be considered a potential diagnosis in patients presenting with MPGN. MPGN encompasses a broad spectrum of underlying diseases, and patients may have aHUS or other hereditary conditions in the background. However, in adult-onset MPGN with an unclear family history, these possibilities are not actively considered, which can be a pitfall. To investigate underlying factors associated with MPGN, genetic testing may help avoid steroid therapy, which can lead to adverse events. Thus, the treatment approach for each case should be thoroughly evaluated and tailored accordingly.
Availability of data and materials
The data that support the results of this study are available on request from the corresponding author. The data are not publicly available because of privacy or ethical restrictions.
References
Gemperle O, Neuweiler J, Reutter FW, Hildebrandt F, Krapf R. Familial glomerulopathy with giant fibrillar (fibronectin-positive) deposits: 15-year follow up in a large kindred. Am J Kidney Dis. 1996;28(5):668–75.
Masabumi Y, Naoto M, Takahiro O, Keisuke S, Wataru K, Kazuhiro N, et al. Clinicopathological analysis of glomerulopathy with fibronectin deposits (GFND): a case of sporadic, elderly-onset GFND with codeposition of IgA, C1q, and fibrinogen. Intern Med. 2013;52(15):1715–20.
Wang T, Bw H. Fibronectin glomerulopathy: a case report and literature review. Nefrogia (Engl Ed). 2021;41(1):74–6.
Mori T, Kazuyoshi H, Motoko C, Shintaro M, Hirofumi N, Eisei S, et al. Comprehensive genetic testing approach for major inherited kidney diseases, using next-generation sequencing with a custom panel. Clin Exp Nephrol. 2017;21(1):63–75.
Ohtsubo H, Taro O, Kandai N, Yutaka T, Akemi S, Katsuhiro A, et al. Identification of mutations in FN1 leading to glomerulopathy with fibronectin deposits. Pediatr Nephrol. 2016;31(9):1459–67.
Castelletti F, Donadelli R, Banterla F, Hildebrandt F, Zipfel PF, Bresin E, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci USA. 2008;105(7):2538–43.
Wu C. Integrin mediated fibronectin matrix assembly. Trends Glycosci Glycotechnol. 1996;43(8):315–25.
Oh GJ, Waldo A, Paez-Cruz F, Gipson PE, Pesenson A, Selewski DT, et al. Steroid-associated side effects in patients with primary proteinuric kidney disease. Kidney Int Rep. 2019;4(11):1608–16.
Noris M, Remuzzi G. Translational mini-review series on complement factor H: therapies of renal diseases associated with complement factor H abnormalities: atypical haemolytic uraemic syndrome and membranoproliferative glomerulonephritis. Clin Exp Immunol. 2008;151(2):199–209.
Goldman BI, Panner BJ, Welle SL, Gross MD, Gray DA. Prednisone-induced sustained remission in a patient with familial fibronectin glomerulopathy (GFND). CEN Case Rep. 2021;10(4):510–4.
Brotman DJ, Girod JP, Posch A, Jani JT, Patel JV, Gupta M, et al. Effects of short-term glucocorticoids on hemo-static factors in healthy volunteers. Thromb Res. 2006;118(2):247–52.
Acknowledgements
The authors would like to thank Dr. Masaaki Takizawa, and Dr. Nagomi Yashiro and the Neurology Department, Respiratory Medicine Department, and Infectious Disease Department for their support in clinical practice
Funding
This work was supported by Grant-in-Aid for Scientific Research (B) (22H03085) to S.U., Grant-in-Aid for Scientific Research (B) (19H03672) and Grant-in-Aid for challenging Exploratory Research (22K19518) to E.S., Grant-in-Aid for Young Scientists (19K17733), Grant-in-Aid for Scientific Research (C) (21K08249) to T.M. from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Contributions
MH and TM wrote the manuscript. TM performed genetic testing and interpreted the genetic data. MH, TM, YH, and YN were responsible for patient care. SM, FA, KS, SI, SN, ES, TR, and SU contributed to the discussion on treatment strategies. TT, ST, and KO contributed to the pathological diagnosis. All authors critically revised the draft for important intellectual content, read, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants provided written informed consent and agreed that their clinical information and DNA could be used in studies to identify genetic risk variants for kidney function. The Institutional Review Board of the Tokyo Medical and Dental University approved this study (Approval no. G2000-080).
Consent for publication
Written informed consent was obtained from the patient for the publication of this case report.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Fig. 1.
Immunofluorescence (IF) staining of the renal biopsy. The results of IF staining showed weak deposition of immunoglobulin (Ig) G, with no evident deposition of other IgA, IgM, C3c, C4, or C1q.
Additional file 2: Supplementary Table.
121 known genes validated in the targeted NGS panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hata, M., Mori, T., Hirose, Y. et al. A case of unexpected diagnosis of fibronectin glomerulopathy with histological features of membranoproliferative glomerulonephritis. BMC Nephrol 25, 25 (2024). https://doi.org/10.1186/s12882-024-03456-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-024-03456-7