
Abstract
Background
Galloway-Mowat syndrome (GAMOS) is a group of rare hereditary diseases by the combination of early onset steroid-resistant nephrotic syndrome (SRNS) and microcephaly with brain anomalies caused by WDR73, LAGE3, OSGEP, TP53RK, TPRKB, GON7, WDR4 or NUP133 mutations.
Case presentation
We present the clinical and genetic features of a two-year-old boy with early nephrotic syndrome, microcephaly, growth retardation hypotonia and hypothyroidism. Genetic testing showed the presence of a canonical-splice mutation in the LAGE3 gene (NM_006014: c.188 + 1C > T). A total of nine female members of the family carried the variant. Seven male members died prematurely, and three of them suffered from nephrotic syndrome, which is consistent with the x-linked gene map of the disease. The overall symptoms of the disease due to the LAGE3 mutation were mild compared to other pathogenic genes.
Conclusion
As far as we know, this is the largest family case of GAMOS2 caused by LAGE3 mutation found so far. We also compared other subtypes of GAMOS. Due to the heterogeneity of the renal phenotype, regular proteinuria screening is recommended for all patients diagnosed with GAMOS.
Similar content being viewed by others


Background
Galloway-Mowat syndrome (GAMOS) is a rare genetic disease that has these typical clinical manifestations, such as microcephaly, central nervous system abnormalities, and early-onset hormone-resistant nephrotic syndrome (steroid-resistant nephrotic syndrome, SRNS). In recent years, with the deepening of disease research, 54 GAMOS families with genetic abnormalities have been reported globally. In addition to kidney and neurological symptoms, clinical phenotypes have been found to include intrauterine growth retardation, optic nerve atrophy, and nystagmus, hearing impairment, craniofacial deformities, abnormal skeletal development of extremities, congenital heart disease and hypothyroidism. 67% of cases died before the age of three [1]. We report on the probands and their families of Galloway-Mowat syndrome caused by a classic splicing mutation in the LAGE3 (L antigen family member3) gene.
Case presentation
A 2-year-old male patient presented to our hospital in September 2019 due to "abnormal urine test found for 11 days". The physical examination of the patient 11 days before admission was found to be positive for urine protein, and the 24-h urine protein was 784 mg. The patient showed growth retardation and delayed development in the past. He raised his head at 6 months, sat at 10 months, walked at 1 year and 8 months, and is now 2 years old with unstable gait and can speak individual monosyllable words. The father suffers from ankylosing spondylitis, the mother has a positive urine protein, and they are not consanguineous. The patient has a sibling who is in good health. Physical examination of the patient on admission: body temperature 36.5 ℃, respiratory rate 27 breaths/min, heart rate 115 beats/min, head circumference 45.3 cm, weight 11.2 kg (10-25th percentile), height 80 cm (below the 3rd percetile);.There are simian lines on both hands.All fingers are short and stubby.The forehead is narrow and slanted. The patient's cardiopulmonary examination is not special. He has weak muscle tone (Fig. 1).Main laboratory results showed on Table 1. Immune set, ANCA-related antibodies, anti-glomerular basement membrane antibodies, four items before blood transfusion, urinary system ultrasound, electroencephalogram, and brainstem auditory evoked potentials were all normal.

The image of the proband. A and B The forehead of the proband is narrow and inclined. C All fingers are short and stubby. There are simian lines on both hands
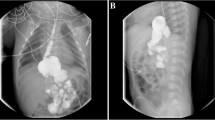
After the child was admitted to the hospital, the examination was completed, and levothyroxine tablets were taken orally to treat hypothyroidism. The proband undergoes kidney biopsy under general anesthesia. The pathological results suggested that 2/25 glomeruli were focal segmental sclerosis. Immunofluorescence showed no deposition of immune complexes. Under the electron microscope, the glomerular foot processes were diffusely fused with microvilli, and no precise electron dense deposits were seen. The patient was diagnosed with focal segmental glomerulosclerosis (FSGS) (Fig. 2).

The image of renal biopsy from the proband. A PASM staining. B MASSON staining glomerular sclerosis. C Foot process fusion under electron microscope)
Combined with the child's medical history and family history, genetically related nephrotic syndrome may be considered. Further investigation of the family history revealed that many male members of the family died prematurely with unknown etiology. One of the male members who died (III-3) had abnormal urine test. The other three male members (IV-5, IV-6 and IV-9) suffered from nephrotic syndrome and died at the age of 3, 5 and 8. In order to clarify the cause, after medical ethics review and the parents of the child signed an informed consent form, 2 mL of the peripheral blood samples of the child and the parents were collected for whole-exome genome sequencing. The results showed that the LAGE3 gene on the X chromosome of the child had a classical splicing site (NM_006014.4:c.188 + 1G > A), which was rated as pathogenic according to ACMG guidelines. This variant is a known pathogenic variant [1]. At present, the professional version of HGMD data only includes 4 variants of the LAGE3 gene, including one classic splice site (c.188 + 1G > A, c.317 + 4A > G), and two missense variant sites (c.316G). > T, c.410 T > C). The LAGE3 gene includes 3 exons. The encoded protein has a functional domain Pcc1 (transcription factor Pcc1). Pcc1 is a transcription factor that functions in regulating genes involved in cell cycle progression and polarised growth [2] (Fig. 3).

Schematic diagram of LAGE3 gene structure and protein domain
The family members of the proband were verified by first-generation sequencing, and it was found that the mutation site was inherited from the mother, and the father and sibling brother were wild type. A total of 9 female members of the family (including the mother of the proband) all carried the mutation site, of which III-2, III-7 and III-8 were positive for urine protein, and IV-7, IV-8 and IV-10 for urine protein feminine. No urine routine was tested in II-2, II-6, and III-4. In addition, I-1 was due to Alzheimer's disease, and I-2 was due to brain tumor. II-3, II-4, and II-5 all died before the age of 5, and the specific cause was unknown. III-3 had abnormal urine test and died at the age of 5. IV-5, IV-6 and IV-9 all suffered from nephrotic syndrome and died at the age of 3, 5 and 8 years old respectively (Fig. 4).

The pedigree of a family with Gallowy-Mowat syndrome caused by splicing mutation of LAGE3.
 indicates female members carrying LAGE3 mutation sites;
indicates female members carrying LAGE3 mutation sites;
 indicates as probands
indicates as probands
The child was discharged after a complete examination. Regular follow-up visits are currently in the outpatient clinic. The child continued to take thyroxine tablets to treat hypothyroidism. At present, there is no abnormality in thyroid function of the patient.The case was not treated with glucocorticoid and immunosuppressant because of the lack of evidence-based medicine.Test and evaluate his urine output, blood pressure, and monitor urine routine, urine protein quantitative, kidney function and other related indicators.
Discussion
Galloway-Mowat syndrome (GAMOS) was first reported in 1968, describing a pair of siblings suffering from the primary nephrotic syndrome-hiatal hernia-microcephaly triad [3].
Recent studies have revealed the important role of gene mutation in the pathogenesis of GAMOS. In 2014, Colin et al. [4] first reported that the loss of WDR73 (WD repeat domain 73) expression can lead to abnormal nuclei of glomerular podocytes, changes in microtubule networks, and cell viability. Braun et al. [1] found that the subunits coded by the four genes LAGE3/OSGEP/TP53RK/TPRKB constitute a highly conserved kinase-endopeptidase and other proteins of small size complex (KEOPS), which is one of the key factors in the pathogenesis of GAMOS. In animal experiments, OSGEP, TPRK or LAGE3 mutations could lead to small head type in the early stage of zebrafish juvenile models. It also resulted in a significant reduction in the length of the cerebral cortex in mouse embryonic models. However, no kidney phenotype had been observed in the existing animal models, and it was speculated that the early lethality masked the appearance of the kidney phenotype. This indicated that the nervous system phenotype in GAMOS might appear earlier than the kidney involvement. Subsequent research by Arrondel.C [5] found that GON7, the fifth gene that constitutes KEOPS, combines with LAGE3 and participates in cell stability and spatial structure arrangement.
The WDR4 gene [6] is a newly discovered and consistent disease gene in recent years, which is located on chromosome 21 and has an autosomal recessive inheritance. In addition to microcephaly, general developmental delay and proteinuria phenotype, there are hypothyroidism. Growth hormone deficiency and other endocrine complications, but the degree of kidney involvement is not serious.
NUP133 gene [7] mutations can also have clinical manifestations similar to GAMOS. The kidney pathology of 3 tested patients showed FSGS. The zebrafish model with this gene knocked out showed microcephaly, neurocytopenia, glomerular hypoplasia and Podocyte foot process fusion, which is similar to human GAMOS signs.
We summarized GAMOS1-5 types (Table 2) that have been reported to have gene mutations, and found several characteristics of the phenotype [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. (1) High phenotypic heterogeneity. (2) Early onset of neurological symptoms. (3) The time and extent of the appearance of kidney phenotype are closely related to the lifespan of patients.
We reported the GAMOS pedigree with the largest number of people discovered so far. The proband had a clear neurological and kidney phenotype, but the overall symptoms were mild. It was speculated that GAMOS2 caused by LAGE3 mutations was a non-early lethal mutation based on family status. In addition to the proband, there were 7 male members of the family who had fatal manifestations in early childhood and childhood. Among them, 4 had abnormal urine tests, 3 had confirmed nephrotic syndrome, and 2 had been treated with glucocorticoids and other drugs. The cause of their death was ESRD and infection. The samples of the 9 female members submitted for examination all carried the same pathogenic site. Among them, 3 female members aged 30–39 years old had proteinuria, and 3 female members aged 6–10 years old showed no abnormalities in urine test. It was considered that the heterozygous kidney phenotype gradually appears with age. The pedigree analysis was consistent with the characteristics of X-linked inheritance caused by LAGE3. Consistent with the report of Braun [1], it was speculated that the fatal male cases in infancy and childhood in this family were hemizygous, and the kidney phenotype IV-7, IV-8 and IV-10 would gradually present with the increase of age.
The diagnosis time of this family is relatively delayed, and we believe that the main factors are as follows: (1) The patient's maternal family lives in remote rural areas, and the family members have a low overall cultural level and lack of knowledge of genetic diseases. There are neither local doctors with relevant expertise nor genetic testing available. (2) There are obvious individual differences in the severity of clinical phenotypes among different subtypes of GAMOS. For example, compared with the reported GAMOS3 and GAMOS4, IV-5, IV-6 and IV-9 in this family are not early fatal cases, which leads to insufficient attention of family members and doctors to the disease, and no further family history is traced.
Conclusion
Kidney complications are the direct cause of most GAMOS deaths, except for sepsis, pneumonia, and severe electrolyte disturbances in a few cases. In view of the above, recommendations for screening and diagnosis of GAMOS include the following [6, 9, 17, 18]. (1) Prenatal screening and: For high-risk pregnant women with suspicious family history, in addition to genetic testing, prenatal ultrasound and fetal nervous system MRI can assist in the diagnosis in the second trimester. (2) Early diagnosis: In the infant stage, patients with abnormal urinary protein, obvious abnormal appearance, obvious family inheritance, and unexplained death of family members in childhood can be considered for genetic testing to determine or exclude the possibility of GAMOS. (3) Early intervention: kidney replacement therapy and supportive care including dialysis can significantly delay the progression of kidney complications. (4) Drug therapy: At present, no confirmed cases of GAMOS use immunosuppressive agents when the kidney phenotype appears. However, with the popularization of genetic testing, patients with single-gene hereditary nephropathy represented by GAMOS, even if the phenotype is SRNS, may have a partial response to immunosuppressive agents, which is expected to be achieved when combined with other therapies to control proteinuria (such as ACEI drugs) Clinical partial or even complete remission will create opportunities for individualized treatment in the future. (5) Close evaluation: Due to the heterogeneity of kidney phenotypes, it is recommended that all patients diagnosed with GAMOS be screened for proteinuria on a regular basis.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Braun DA, et al. Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly[J]. Nat Genet. 2017;49(8):1529–38.
Kisseleva-Romanova E, et al. Yeast homolog of a cancer-testis antigen defines a new transcription complex[J]. EMBO J. 2006;25(15):3576–85.
Galloway WH, Mowat AP. Congenital microcephaly with hiatus hernia and nephrotic syndrome in two sibs[J]. J Med Genet. 1968;5(4):319–21.
Colin E, et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome[J]. Am J Hum Genet. 2014;95(6):637–48.
Arrondel C, et al. Defects in t6A tRNA modification due to GON7 and YRDC mutations lead to Galloway-Mowat syndrome[J]. Nat Commun. 2019;10:3967–80.
Braun DA, et al. Mutations in WDR4 as a new cause of Galloway-Mowat syndrome[J]. J Med Genet. 2018;176(11):2460–5.
Fujita A, et al. Homozygous splicing mutation in NUP133 causes Galloway-Mowat syndrome[J]. Neurology. 2018;4(6):814–28.
Ben-Omran T, et al. Nonsense mutation in the WDR73 gene is associated with GallowayMowat syndrome[J]. J Med Genet. 2015;52(4):381–90.
Jinks RN, et al. Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73[J]. Brain. 2015;138(6):2173–90.
Rosti RO, et al. Extending the mutation spectrum for Galloway-Mowat syndrome to include homozygous missense mutations in the WDR73 gene[J]. Am J Med Genet Part A. 2016;170A(4):992–8.
Wang P, et al. Nephrological and urological complications of homozygous c.974G>A (p.Arg325Gln) OSGEP mutations[J]. Pediatr Nephrol. 2018;33(11):2201-4.
Edvardson S, et al. TRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and kidney tubulopathy[J]. Eur J Hum Genet. 2017;25(3):545–51.
Al-Rakan MA, Abothnain MD, Alrifai MT, Alfadhel M. Extending the ophthalmological phenotype of Galloway-Mowat syndrome with distinct retinal dysfunction: a report and review of ocular findings[J]. BMC Ophthalmol. 2018;18(6):4–7.
Hyun HS, et al. A familial case of Galloway-Mowat syndrome due to a novel TP53RK mutation: a case report[J]. BMC Med Genet. 2018;19(7):131–7.
Bullich G, et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases[J]. Kidney Int. 2018;94(7):363–71.
Chen CP, et al. Galloway-Mowat syndrome: prenatal ultrasound and perinatal magnetic resonance imaging findings[J]. Taiwan J Obstet Gyneco. 2011;50(52):212–6.
Mubarak M, et al. Early onset nephrotic syndrome with dysmorphic facies and microcephaly[J]. J Nephropathol. 2015;4(3):101–4.
Cil O, et al. Monogenic Causes of Proteinuria in Children[J]. Front Med. 2018;5(3):1–10.
Acknowledgements
We are grateful to the patient and their families for their participation.We thank Liwan Wei of Chigene who helped in the revision process of the article. We thank Lisa Guo of Genokon for her helpful advices during the gene detection.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
CY was a major contributor in writing the manuscript. YY and YY conducted data collection and followed up of patient and families. RJ conducted genetic analyses.BHT planned and supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the First affiliated Hospital of Xiamen university’s Institutional ethics committee (XMFHIIT-2022SL021). The parents of the patient and families provided informed consent to participate in this study.
Consent for publication
The parents of the patient provided informed consent to publish this case report.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Yang, Y., Yang, Y. et al. Diagnosis delay a family of Galloway-Mowat Syndrome caused by a classical splicing mutation of Lage3. BMC Nephrol 24, 29 (2023). https://doi.org/10.1186/s12882-022-03000-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-022-03000-5