
Abstract
Background
Familial renal glucosuria (FRG) is characterized by persistent glucosuria without other impairments of tubular function in the presence of normal serum glucose. SGLT2, which is almost exclusively expressed in the kidney, accounts for most of the glucose reabsorption. Recently, some studies have confirmed that SLC5A2 mutations are responsible for the pathogenesis of familial renal glucosuria, but FRG cases are still rare. Furthermore, there are a few reports about splice-site mutations in previous studies, but the effect of these variants at the mRNA level has hardly been verified.
Methods
Ten patients were recruited in our renal division because of persistent glucosuria, and clinical data of the patients and their family members were recorded as much as possible. The entire coding region and adjacent intronic segments of SLC5A2 were sequenced in FRG patients and their relatives. Permanent growing lymphoblastoid cell lines from FRG patients were established to better preserve genetic information.
Results
A total of nine different mutations were identified: IVS1-16C > A, c.305C > T/p.(A102V), c.395G > A/p.(R132H), c.736C > T/p.(P246S), c.886(−10_-31)delGCAAGCGGGCAGCTGAACGCCC, c.1152_1163delGGTCATGCTGGC/p.(Val385_Ala388del), c.1222G > T/p.(D408Y), c.1496G > A/p.(R499H) and c.1540C > T/p.(P514S); two novel mutations in SLC5A2, c.1222G > T/p.(D408Y) and c.1496G > A/p.(R499H), were identified in the Chinese FRG pedigrees. Ten individuals with heterozygous or compound heterozygous variants had glucosuria in the range of 3.1 to 37.6 g/d.
Conclusion
We screened ten additional Chinese FRG pedigrees for mutations in the SLC5A2 gene and found nine mutations, including two novel mutations. Most variants were private, but IVS1-16C > A and c.886(−10_-31) del may be high frequency splice-site mutations that could be preferentially screened when variants cannot be found in the SLC5A2 exon. Furthermore, we successfully established a permanent growing lymphoblastoid cell line from patients with FRG, which could facilitate further studies of the SLC5A2 gene. The current study provides a valuable clue for further research on the molecular mechanism of SGLT2.
Similar content being viewed by others


Background
Familial renal glycosuria (FRG) is characterized by persistent glycosuria with normal blood sugar concentrations and without any other impairment of tubular function [1]. The main reabsorptive mechanism for D-glucose in the kidney involves a lower affinity, high capacity Na(+)/glucose cotransporter 2 (SGLT2), which is located in the S1 segment of the early proximal convoluted tubule, and a Na(+) and glucose coupling ratio of 1:1 [2]. The SLC5A2 gene was mapped to 16p11.2 [3]. Recently, some published studies have confirmed that SLC5A2 mutations are responsible for FRG patients [4,5,6,7,8,9,10,11,12,13,14,15,16]. In some of these studies, FRG was considered an autosomal recessive disorder [7,8,9,10,11]. In others, it was considered a codominant trait with variable penetrance [5, 6]. In our previous studies, the inheritance of renal glucosuria was best described as codominant with a variable penetrance in relation to the compensatory capacity of wild-type [12, 14, 15]. In long-term follow-up studies, the outcome of FRG patients is very good [5, 17]. SGLT2 inhibitors are designed to improve the condition of diabetes without increasing the risk of weight gain or hypoglycemia. SGLT2 has been the subject of particular attention in the search for potential new drugs for the treatment of diabetes [18]. Here, we describe ten patients with glucosuria of variable severity and nine SLC5A2 mutations. Furthermore, in previous reports, the effect of splice-site variants was rarely verified. We established a permanent growing lymphoblastoid cell line to verify the effect of splice-site variants from previous studies [12].
Methods
Patients with FRG were diagnosed by persistent glycosuria in the presence of a normal serum glucose concentration and no other impairments of tubular function or any other type of renal disease. Ten unrelated FRG patients and their families were investigated as much as possible. The age, sex, serum creatinine, urine protein excretion, glucosuria excretion and other clinical manifestations in all patients were recorded. Fifty-five healthy Chinese individuals were included as controls in our study.
Genomic DNA was extracted by a salting out procedure from peripheral white blood cells from whole blood samples [19]. The products of polymerase chain reaction (PCR) were sequenced, and the genomic DNA reference sequence of SLC5A2 (NG_012892.1, Gene ID: 6524) and protein reference sequence of SGLT2 (NP_003032.1) were acquired from the Entrez gene and protein databases, respectively. In the analysis of variants, the entire coding region and adjacent intronic segments of SLC5A2 were sequenced in family members as much as possible, and the variants were confirmed by bidirectional sequencing. The set of primers used was previously reported [11]. We established a permanent growing lymphoblastoid cell line from patients with FRG as previously reported [12, 20].
A total of 110 control chromosomes were tested by sequencing or polymerase chain reaction-restriction fragment length polymorphism (PCR–RFLP) to rule out common polymorphisms. Furthermore, three databases, including the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/), GnomAD v3 and GnomAD v2.1.1 (http://gnomad.broadinstitute.org), were used to further eliminate polymorphisms.
Amino acid substitutions were evaluated using the in silico prediction programs SIFT and PolyPhen-2. In addition, a comparative analysis of multiple amino acid sequences of SGLT2 was performed in different species by multiple sequence alignments of DNAMAN Version 6. The aligned reference sequences of Homo sapiens (NP_003032.1), Pan troglodytes (XP_009428973.2), Macaca mulatta (XP_001113206.3), Bos taurus (NP_976236.1), Rattus norvegicus (NP_072112.2), Mus musculus (NP_573517.1), Danio rerio (NP_998091.1) and Xenopus tropicalis (XP_002940641.2) were used to evaluate the evolutionary conservation.
Results
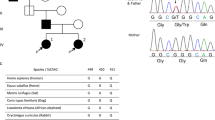
All ten patients met the diagnostic criteria of FRG. These patients and their families did not have any other tubular dysfunctions or any other type of renal disease. A total of nine different mutations were identified: IVS1-16C > A, c.305C > T/p.(A102V), c.395G > A/p.(R132H), c.736C > T/p.(P246S), c.886(−10_-31) delGCAAGCGGGCAGCTGAACGCCC, c.1152_1163delGGTCATGCTGGC/p.(Val385_Ala388del), c.1222G > T/p.(D408Y), c.1496G > A/p.(R499H) and c.1540C > T/p.(P514S); two novel mutations in SLC5A2, c.1222G > T/p.(D408Y) and c.1496G > A/p.(R499H), were identified in the Chinese FRG pedigrees (Fig. 1). By PCR–RFLP testing or sequencing, these variants were not found in one hundred and ten chromosomes derived from the fifty-five healthy unrelated individuals (Table 1). Because allele frequencies for the observed variants in the Chinese population are still unknown, extremely low allele frequencies of these variants were alternatively obtained in East Asian patients from the ExAC and gnomAD databases (Table 2).

Ten familial renal glucosuria pedigrees carry SLC5A2 variants. A total of nine different mutations were identified: IVS1-16C > A, c.305C > T/p.(A102V), c.395G > A/p.(R132H), c.736C > T/p.(P246S), c.886(−10_-31)delGCAAGCGGGCAGCTGAACGCCC, c.1152_1163delGGTCATGCTGGC/p.(Val385_Ala388del), c.1222G > T/p.(D408Y), c.1496G > A/p.(R499H) and c.1540C > T/p.(P514S); two novel mutations in SLC5A2, c.1222G > T/p.(D408Y) and c.1496G > A/p.(R499H), were identified in the Chinese FRG pedigrees. Ten individuals were heterozygous or compound heterozygous for an SGLT2 mutation, resulting in glucosuria. The missense variants were predicted to be possibly damaging by PolyPhen-2
The identified missense variants are highly conserved in SGLT2 homologs in multiple species (Fig. 2). By PolyPhen-2 [21], all of these missense variants were predicted to be “probably damaging” (Fig. 1, Table 3). Consistent with PolyPhen-2, five missense variants, c.305C > T/p.(A102V), c.395G > A/p.(R132H), c.1222G > T/p.(D408Y), c.1496G > A/p.(R499H) and c.1540C > T/p.(P514S), were predicted by SIFT to “affect protein function”, but the c.736C > T/p.(P246S) variant was predicted to be “tolerated” by SIFT, which was different from the PolyPhen-2 prediction (Table 3).

Multiple sequence alignment of the SGLT2 protein from different species. Six conserved amino acids in SGLT2 were identical among different species and are highlighted
Glucosuria ranged from 3.1 to 37.6 g/d in ten patients with SLC5A2 heterozygous or compound heterozygous variants. Some of the family members with heterozygous variants had increased glucose excretion (Table 1). In these families, inheritance of FRG shows characteristics of a codominant trait with variable penetrance. Most variants were private, but the IVS1-16C > A and c.886(−10_-31) del variants were reported in several unrelated pedigrees from different ethnic origins in our and previous studies [12,13,14,15,16, 22, 23].
Discussion
Glucose, mainly from carbohydrates, is the fuel that provides energy for human activities. The kidneys reabsorb nearly 180 g of glucose filtered daily to keep blood glucose in the normal range. In previous studies, familial renal glycosuria was characterized by persistent glycosuria, and the SGLT2 protein was found to be mainly responsible for the reabsorption of urinary glucose in renal tubules [1, 24, 25]. Therefore, it was speculated that SLC5A2 gene mutations lead to familial renal glycosuria. The first report of an SLC5A2 mutation in FRG was presented in 2000 [26]. Recently, a series of studies have confirmed that SLC5A2 mutations are indeed responsible for FRG [4,5,6,7,8,9,10,11,12,13,14,15,16, 22, 23]. In our previous and current studies, fourteen novel variants in SLC5A2 were identified in twenty-two Chinese renal glucosuria families and confirm previous observations that most variants were private mutations. With an increasing number of FRG patients being found, some variants, such as 294C > A, IVS1-16C > A, c.886(−10_-31) del, and c.1540G > T, did not occur rarely in our and previous studies [12,13,14,15,16, 22, 23]. However, these variants are difficult to regard as hotspot mutations because they were found in SLC5A2 with a relatively dispersed distribution. Whereas the IVS1-16C > A and c.886(−10_-31) del variants are reported in several unrelated pedigrees of different ethnic origins, these two splice-site variants might be preferentially screened in FRG patients when the other variants cannot be found in the SLC5A2 exon. Furthermore, specific novel primers should be developed to check for the presence of the observed splice site variants in genomic DNA in future studies.
In the current study, a total of nine different mutations in SLC5A2 were identified in the Chinese FRG pedigrees. None of these variants were found in one hundred and ten chromosomes from healthy unrelated individuals. In addition, the allele frequencies for these variants were extremely low in East Asian populations. For the identified missense variants, five variants, c.305C > T/p.(A102V), c.395G > A/p.(R132H), c.1222G > T/p.(D408Y), c.1496G > A/p.(R499H) and c.1540C > T/p.(P514S), were highly conserved in SGLT2 homologs in multiple species and were predicted to be “probably damaging” or to “affect protein function” by PolyPhen-2 and SIFT. Only the variant of c.736C > T/p. (P246S) was an exception, and was predicted by SIFT to be “tolerated”, but this variant was reported in previous studies [12, 27] and was confirmed by having a significantly lower glucose transport capacity in cultured cells [12]. Therefore, based on the extremely low allele frequencies of these mutations, highly conservative predictions from biological software and previous studies, it can be safety speculated that these variants are not common polymorphisms and are pathogenic mutations.
In previous studies, many heterozygous individuals presented with mild glucosuria (< 10 g/d), while homozygous or compound heterozygous patients usually present with severe renal glucosuria over 10 g/d [4, 5]. The heterozygosity of SLC5A2 mutations, no matter what kind of mutation (such as nonsense, splice-site, and missense mutations), can lead to mild glucosuria. Consistent with previous research, six individuals were heterozygous for SLC5A2 variants resulting in mild glucosuria (< 10 g/d), and one compound heterozygous patient from Family II had severe renal glucosuria (37.6 g/d) in the current study. It is very interesting that three heterozygous patients from Families II, V, and X resulted in severe renal glucosuria (> 10 g/d), and further studies are needed to uncover the related regulatory mechanism.
In the current study, an autosomal codominant trait with variable penetrance inheritance was found in FRG families. In our previous studies, we found that the inheritance of renal glucosuria should be described as codominant with a variable penetrance in relation to the compensatory capacity of wild-type [14, 15]. Different modes of penetrance inheritance may be decided by different sites or other special regulatory mechanisms. Thus, reporting mutations is crucial not only for unraveling critical residues in the protein but also for obtaining useful information to identify potential new targets for the treatment of diabetes.
Renal biopsy is not obligatory for FRG patients; therefore, SLC5A2 cDNA from the kidney is almost impossible to obtain. Although there are a few reports about splice-site variants [4, 5, 22, 23], the effect of splice-site variants is very difficult to verify in cDNA. We searched through NCBI GEO profiles and found that the SGLT2 protein can be expressed in peripheral white blood cells and lymphocytes. However, due to the limited expression and lifespan of these cells, new blood sampling is necessary via repeatedly drawing blood for reexamination. This might be difficult or even impossible if patients were not available for different reasons. In 1986, a routine method for the establishment of permanent growing lymphoblastoid cell lines was reported [20]. In a previous report, the Epstein–Barr virus genome not only persists as a plasmid with 5–800 copies per cell in most cell lines but also integrates into the host DNA and has been described for a few cell lines [18]. However, there have been no reports on establishing lymphoblastoid cell lines from FRG families in previous studies. We successfully established a permanent growing lymphoblastoid cell line from patients with FRG and successfully verified the effects of splice-site mutations at the cDNA level [12]. Although the integration into the host DNA may affect genetic information, the integration of the Epstein–Barr virus in lymphoblastoid cell lines is nonrandom [28]. The viral integration sites included 1p, 1q, 2q, 3p, 3q, 4q, 5q, 6q, 7p, 7q, 9q, 11p, 14q and 15q. No viral integration occurred in chromosomes 16–22 or the sex chromosomes [28,29,30]. Because the SLC5A2 gene was mapped to 16p11.2 and there were no Epstein–Barr virus gene sequences in the cDNA sequencing results, we confirmed that a permanent growing lymphoblastoid cell line from FRG patients was successfully established. In the current study, we found two splice site variants: IVS1-16C > A and c.1152-63del. However, the effect of these two splice-site variants has been verified in previous studies [13, 22]. Therefore, we did not retest the effect of splice-site variants in cDNA in the current study. However, we still believe that the method for establishing permanent growing lymphoblastoid cell lines in patients with FRG is useful to maintain genetic information about SLC5A2 and more easily verify the effect of splice-site variants in cDNA.
In previous studies, the variant frequency of c.886(−10_-31) del in the Chinese population was as high as 32% [22, 23]. Therefore, we rescreened the observed splice site variants in every patient from the twenty-two Chinese renal glucosuria families that were found in our previous and current studies. Finally, except for splice site mutations that were previously found, no additional splice site variants were discovered in these renal glucosuria families.
Conclusions
In conclusion, we screened ten additional Chinese FRG pedigrees and found nine SLC5A2 mutations, including two novel mutations. The variants IVS1-16C > A and c.886(−10_-31) del, which had high frequencies, could be preferentially screened in FRG patients when the variants cannot be found in exons. In addition, we established a permanent growing lymphoblastoid cell line from patients with FRG, which could facilitate further studies of the SLC5A2 gene at the cDNA level. In short, our study provides valuable clues for further studies of the SGLT2 molecular mechanism and potential targets for the further development of anti-diabetes drugs.
Availability of data and materials
Additional data used/generated that is not present in the manuscript is available from the corresponding author upon reasonable request.
Abbreviations
- EB:
-
Epstein–Barr virus
- FRG:
-
Familial renal glucosuria
- GLUT:
-
Glucose transporter
- PCR:
-
Polymerase chain reaction
- PCR–RFLP:
-
Polymerase chain reaction-restriction fragment length polymorphism
- SGLT2:
-
Sodium-glucose cotransporter 2
- SLC5:
-
Sodium-glucose cotransporter family
- SLC5A2 :
-
Sodium-glucose cotransporter 2 gene
References
Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol Renal Physiol. 2001;280:F10–8.
Kanai Y, Lee WS, You G, Brown D, Hediger MA. The human kidney low affinity Na(+)/glucose cotransporter SGLT2: delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest. 1994;93:397–404.
Wells RG, Mohandas TK, Hediger MA. Localization of the Na+/glucose cotransporter gene SGLT2 to human chromosome 16 close to the centromere. Genomics. 1993;17:787–9.
Calado J, Sznajer Y, Metzger D, Rita A, Hogan MC, Kattamis A, et al. Twenty-one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol Dial Transplant. 2008;23:3874–9.
Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, et al. Analysis of the SGLT2 gene in patients with renal Glucosuria. J Am Soc Nephrol. 2003;14:2873–82.
Calado J, Loeffler J, Sakallioglu O, Gok F, Lhotta K, Barata J, et al. Familial renal glucosuria: SLC5A2 mutation analysis and evidence of salt-wasting. Kidney Int. 2006;69:852–5.
Van den Heuvel LP, Assink K, Willemsen M, Monnens L. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet. 2002;111:544–7.
Francis J, Zhang J, Farhi A, Carey H, Geller DS. A novel SGLT2 mutation in a patient with autosomal recessive renal glucosuria. Nephrol Dial Transplant. 2004;19:2893–5.
Calado J, Soto K, Clemente C, Correia P, Rueff J. Novel compound heterozygous mutations in SLC5A2 are responsible for autosomal recessive renal glucosuria. Hum Genet. 2004;114:314–6.
Kleta R, Stuart C, Gill FA, Gahl WA. Renal glucosuria due to SGLT2 mutations. Mol Genet Metab. 2004;82:56–8.
Magen D, Sprecher E, Zelikovic I, Skorecki K. A novel missense mutation in SLC5A2 encoding SGLT2 underlies autosomal-recessive renal glucosuria and aminoaciduria. Kidney Int. 2005;67:34–41.
Yu L, Hou P, Lv JC, Liu GP, Zhang H. Novel SLC5A2 variants contribute to renal Glucosuria in Chinese families: abnormal expression and dysfunction of variant SLC5A2. Hum Mutat. 2015;36:79–86.
Yu L, Lv J-C, Zhou X-j, Zhu L, Hou P, Zhang H. Abnormal expression and dysfunction of novel SGLT2 mutations identified in familial renal glucosuria patients. Hum Genet. 2011;129:335–44.
Yu L, Hou P, Lv JC, Liu GP, Zhang H. A novel sodium-glucose co-transporter 2 gene (SGLT2) mutation contributes to the abnormal expression of SGLT2 in renal tissues in familial renal glucosuria. Int Urol Nephrol. 2014;46:2237–8.
Yu L, Hou P, Liu GP, Zhang H. A novel SLC5A2 mutation contributes to familial renal glucosuria: abnormal expression in renal tissues. Experimental and Therapeutic Medicine. 2016;12:649–52.
Yu L, Xu Q, Ping H, Hong Z. Decreased expression and function of sodium-glucose co-transporter 2 from a novel C-terminal mutation: a case report. BMC Nephrol. 2016;17:31.
Scholl-Burgi S, Santer R, Ehrich JH. Long-term outcome of renal glucosuria type 0: the original patient and his natural history. Nephrol Dial Transplant. 2004;19:2394–6.
Isaji M. Sodium-glucose cotransporter inhibitors for diabetes. Curr Opin Investig Drugs. 2007;8:285–92.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
Heidemarie N. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986;73:320–6.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Zhao X, Cui L, Lang Y, Liu T, Lu J, Wang C, et al. A recurrent deletion in the SLC5A2 gene including the intron 7 branch site responsible for familial renal glucosuria. Sci Rep. 2016;6:33920.
Wang S, Zhao X, Zhang R, Wang C, Han Y, Shao L. Identification of ten novel SLC5A2 mutations and determination of the renal threshold for glucose excretion in Chinese patients with familial renal glucosuria. Clin Chim Acta. 2019;490:102–6.
Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med. 2007;261:44–52.
Wright EM, Turk E. The sodium/glucose cotransport family SLC5. Pflugers Arch. 2004;447:510–8.
Santer R, Kinner M, Schneppenheim R. The molecular basis of renal glucosuria: mutations in the gene for a renal glucose transporter (SGLT2). J Inherit Metab Dis. 2000;23:178.
Lee H, Han KH, Park HW, Shin JI, Kim CJ, Namgung MK, et al. Familial renal glucosuria: a clinicogenetic study of 23 additional cases. Pediatr Nephrol. 2012;27:1091–5.
Lestou VS, De Braekeleer M, Strehl S, Ott G, Gadner H, Ambros PF. Non-random integration of Epstein-Barr virus in lymphoblastoid cell lines. Genes Chromosomes Cancer. 1993;8:38–48.
Wuu KD, Chen YJ, Wuu SW. Frequency and distribution of chromosomal integration sites of the Epstein-Barr virus genome. J Formos Med Assoc. 1996;95:911–6.
Gao J, Luo X, Tang K, Li X, Li G. Epstein-Barr virus integrates frequently into chromosome 4q, 2q, 1q and 7q of Burkitt's lymphoma cell line (Raji). J Virol Methods. 2000;136:193–9.
Acknowledgements
Not applicable.
Funding
This study was funded by the National Natural Science Foundation of China (grant nos. 81660117 and 81160087), the Natural Science Fund of Inner Mongolia Autonomous Region (grant no. 2018MS08008), and the 2015 Science and Technology Planning Project of Inner Mongolia Autonomous Region (grant no. 201502107). The funders had no role in designing the study, in collecting, analyzing, and interpreting the data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YL collected the clinical information and drafted the manuscript. WM supported the data collection, interpretation of the data, and writing of the manuscript. YL and HP carried out genetic studies and evaluated the mutants. ZH reviewed the draft and made critical modifications. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Medical Ethics Committee of Inner Mongolia People’s Hospital (Hohhot, China). Written informed consent was obtained from all participants prior to participating in the study.
Consent for publication
Written informed consent for publication was obtained from the participants.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yu, L., Wu, M., Hou, P. et al. SLC5A2 mutations, including two novel mutations, responsible for renal glucosuria in Chinese families. BMC Nephrol 21, 69 (2020). https://doi.org/10.1186/s12882-020-01725-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-020-01725-9