
Abstract
Background
Prune belly syndrome (PBS) is a rare, multi-system congenital myopathy primarily affecting males that is poorly described genetically. Phenotypically, its morbidity spans from mild to lethal, however, all isolated PBS cases manifest three cardinal pathological features: 1) wrinkled flaccid ventral abdominal wall with skeletal muscle deficiency, 2) urinary tract dilation with poorly contractile smooth muscle, and 3) intra-abdominal undescended testes. Despite evidence for a genetic basis, previously reported PBS autosomal candidate genes only account for one consanguineous family and single cases.
Methods
We performed whole exome sequencing (WES) of two maternal adult half-brothers with syndromic PBS (PBS + Otopalatodigital spectrum disorder [OPDSD]) and two unrelated sporadic individuals with isolated PBS and further functionally validated the identified mutations.
Results
We identified three unreported hemizygous missense point mutations in the X-chromosome gene Filamin A (FLNA) (c.4952 C > T (p.A1448V), c.6727C > T (p.C2160R), c.5966 G > A (p.G2236E)) in two related cases and two unrelated sporadic individuals. Two of the three PBS mutations map to the highly regulatory, stretch-sensing Ig19–21 region of FLNA and enhance binding to intracellular tails of the transmembrane receptor β-integrin 1 (ITGβ1).
Conclusions
FLNA is a regulatory actin-crosslinking protein that functions in smooth muscle cells as a mechanosensing molecular scaffold, transmitting force signals from the actin-myosin motor units and cytoskeleton via binding partners to the extracellular matrix. This is the first evidence for an X-linked cause of PBS in multiple unrelated individuals and expands the phenotypic spectrum associated with FLNA in males surviving even into adulthood.
Similar content being viewed by others


Background
Prune Belly Syndrome (PBS), also known as Eagle-Barrett or Triad Syndrome (MIM#100100) is a congenital myopathy with highly variable multisystem phenotypic severity, affecting 1 in 25,000 individuals [1,2,3]. The classic triad defining PBS, which almost exclusively affects males, includes 1) the wrinkled, prune-like ventral abdominal skin with underlying flaccid hypoplastic skeletal muscle, 2) urinary tract dilation including megacystis and megaureter with poorly contractile smooth muscle, and 3) bilateral intraabdominal cryptorchidism. However, the PBS phenotype has been categorized into isolated PBS, syndromic PBS, and PBS-plus, based on the absence or presence of genetically defined syndromes or additional malformations outside the classic triad, respectively [2]. Megacystis +/− oligohydramnios is prenatally diagnosed by ultrasound in most cases of PBS with premature birth in 43% and neonatal death in 23–45% [1, 4]. Survivors typically have normal cognition but battle multi-organ dysfunction with ~ 50% developing chronic renal insufficiency or end-stage renal disease [4,5,6,7]. It has been estimated that there are 1494 males under the age of 18 years living with PBS in the US [8].
Historically, the cause of PBS was attributed to a mechanical in utero bladder outlet obstruction. However, a true urethral obstruction is observed in only 10–20% of PBS autopsy cases and does not account for situations where significant abdominal muscular hypoplasia is observed with only a mild urogenital defect [9]. Thus, an alternative etiological hypothesis is that PBS is a consequence of a mesodermal field defect of unknown cause [10]. Most PBS cases are sporadic and no environmental factors have been implicated as causal. Rare PBS case reports have noted abnormal karyotypes, including ring X chromosome, trisomy 13, 18 and 21 [11,12,13,14,15,16,17,18]. There are three pieces of evidence that suggest a genetic basis influenced by X-linked recessive or sex-limited autosomal recessive genetic factor(s) [19]. First, rare concordant PBS twin gestations have been reported [20,21,22]. Second, > 95% of cases are male. Lastly, there have been 12 published multiplex pedigrees without causal genes identified in most [19,20,21, 23,24,25,26,27,28,29,30,31]. More recently, five autosomal genes, including CHRM3, HNF1β, ACTA2, ACTG2 and STIM1, have been reported with potentially causal DNA variants, including structural, copy number, and single nucleotide variants, however these genes each only account for one or two PBS cases or one PBS multiplex consanguineous kindred [32,33,34,35,36,37,38]. Moreover, none of the currently suggested candidate genes fit an X-linked recessive mode of inheritance and functional data is lacking for many of these candidate genetic variants.
In this report, we identify four PBS-affected individuals, two sporadic and two from a multiplex kindred, to have mutations in the X-chromosome gene, FLNA. FLNA encodes for a large actin-binding scaffolding protein that functions in diverse cellular processes including cell migration, wound healing and organ development [39, 40]. Mutations in FLNA underlie a wide spectrum of human disease phenotypes broadly termed the filaminopathies that include periventricular nodular heterotopia (PVNH), the otopalatodigital spectrum disorders (OPDSD) which includes Melnick-Needles Syndrome (MNS) and frontometaphyseal dysplasia (FMD) as well as X-linked cardiac valvular dystrophy (XCVD), among others. Given the X-linkage, phenotypic severity is highly variable in females depending on the exact FLNA variant and the impact of skewed X inactivation. In contrast, most male FLNA mutation carriers, especially those with OPD2 and MNS, die in utero or early antenatally [41, 42]. We now add a new cohort of males surviving even into adulthood with FLNA missense mutations, expanding the spectrum of FLNA phenotypes to include males with syndromic PBS with OPDSD or those only with isolated PBS.
Methods
Study subjects
Internationally, we prospectively enrolled individuals with PBS and their family members in our IRB-approved Pediatric Genitourinary DNA Repository starting in 2001. Informed consent was obtained from all individuals included in the study which was approved by the institutional review board at UT Southwestern Medical Center and all procedures followed were in accordance with the ethical standards of the relevant committees on human experimentation. Patients’ medical records were retrospectively reviewed and in person and/or telephone interviews were performed to obtain medical, surgical and family history. Medical photographs were made and reviewed by a clinical geneticist (AES). Each individual with PBS was assigned a PBS severity score using the RUBACE (R: renal, U: ureter, B: bladder/outlet, A: abdominal wall, C: cryptorchidism, E: extra-genitourinary) phenotyping scoring system developed by our group to better grade disease severity and categorize patients into isolated PBS, syndromic PBS or PBS-Plus groups [2].
Whole exome sequencing
Lymphocyte genomic DNA was extracted according to standard procedures from participants using the Puregene DNA isolation kit (Gentra/Qiagen) or from saliva (Oragene). Paired-end Whole Exome Sequencing (WES) was performed at the UTSW McDermott Next Generation Sequencing Core using the Illumina HiSeq2500. Library preparation was done using the Illumina SureSelect DNA Sample prep kit and capture with the Illumina SureSelect Exome Enrichment kit. Data processing and analysis was performed by the UTSW McDermott Center Bioinformatics group. Adaptor removal and sample demultiplexing was done using CASAVA, BWA was used for alignment to the human genome (GRCh37/hg19), mapped reads were processed, sorted and underwent duplicate removal using Samtools and PICARD, and GATK was used for quality control, including realignment around insertions and deletions and base quality score recalibration. Variant calling was performed using training sets from data from the 1000 Genomes Project, Omni 2.5 M SNP microarray and HapMap phase 3.3. Variant call files were filtered to exclude those with a minor allele frequency (MAF) too high to account for PBS from public databases including, ExAC, gnomAD, 1000 genomes, (ExAC AF < 0.005 for homozygous variants and ExAC AF < 0.00005 for heterozygous or hemizygous variants) and only functional variants (predicted to alter mRNA splicing or amino acid sequences) were included for further analysis.
Gene expression studies
mRNA expression was assessed across normal adult human pooled RNA samples (BioChain) using qPCR following standard methods. Briefly, 1 μg of RNA was reverse transcribed (BioRad iScript cDNA synthesis kit) with oligo (dT) and random hexamer primers. Gene specific primers (FLNA: for 5′- CTGTCACAGTGTCAATCGGAGG and rev 5′- TCGAAAGTGCCGTCCTCATT; ITGB1: for 5′- CCTACTTCTGCACGATGTGATG and rev 5′ - CCTTTGCTACGGTTGGTTACATT) were used to amplify mRNA using KAPA SYBR green mix on the CFX Connect Real Time System (BioRad). Differential gene expression was calculated via the delta delta CT method and normalized to GAPDH and all samples were run in triplicate (n = 3). Significance was calculated by t-test using Graph Pad Prism 7.03 software.
Immunohistochemistry and histology: Bladder biopsy, obtained from consenting normal pediatric individuals at the time of bladder surgery to correct refluxing ureters (n = 4), was fixed in 10% neutral buffered formalin and routinely processed for paraffin embedding. Briefly, tissues were dehydrated in sequentially increasing ethanol concentrations ending with xylene and infiltrated with paraffin. Tissues were embedded in paraffin and sectioned at 4uM. Hematoxylin and eosin staining was used to assess tissue morphology. Immunohistochemistry was performed (FLNA #HPA01115 (Sigma) and ITGβ1 #9699 (Cell Science Technology)) at the UTSW Tissue Management Shared Resource. Staining was automated on the Dako Autostainer Link 48 system to ensure identical staining conditions. Slides were scanned using the Hammamatsu nanozoomer 2.0.
Binding assays
Full length human FLNA with C-terminal GFP in pcDNA3 (Calderwood Lab, Yale University) was used as template to introduce all identified FLNA mutations with the QuikChange II XL kit (Agilent) and verified by Sanger sequencing of the complete FLNA insert to rule out any additional mutations. For analysis of binding of full length FLNA to integrin, CHO (Chinese Hamster Ovary) cells were transiently transfected with FLNA plasmids using Lipofectamine 3000 (Invitrogen). Binding was assessed to purified integrin tails, β1 wild type, β1 Y788A, and β7 (Calderwood Lab, Yale University) following protocols previously described [43] with the modifications that 10 mM imidazole was included in buffer XT and the beads were washed three times.
Results
Clinical findings of PBS patients (Figs. 1 and 2, supplemental figs. 1 and 2)
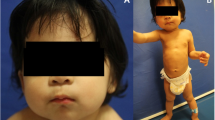
Subjects 1 and 2 (Fig. 1) are maternally shared PBS-affected half-brothers from Pedigree 1 (Fig. 2b) that present with syndromic PBS (PBS with previously undiagnosed OPDSD).

Thoraco-abdominal and Facial Photographs of 4 PBS affected individuals with FLNA mutations. A1-A2) Pedigree 1 Subject 1 (p.C2160R) has a RUBACE severity score of 22 and syndromic PBS with additional OPDSD features including prominent supraorbital ridge and micrognathia (A2). B1-B2) Pedigree 1 Subject 2 (p.c2160R) has a RUBACE severity score of 24 as well as syndromic PBS with OPDSD phenotypic features including prominent supraorbital ridge, micrognathia, facial asymmetry (B2). He additionally has Pierre Robin Sequence. C1-C2) Pedigree 2 Subject 3 (p.A1448V) has a RUBACE severity score of 14 (isolated PBS). No strong OPDSD craniofacial features are noted (C2). D1-D2) Pedigree 3 Subject 4 (p.G2236E) has a RUBACE severity score of 13 (isolated PBS). No strong OPDSD craniofacial features are noted (D2)

Pedigrees and FLNA mutations in PBS patients. a FLNA dimer is composed of an N-terminal actin binding domain (ABD), 24 immunoglobulin (Ig) repeats with two calpain-sensitive hinges separating the IgFLNA repeat domains into Rod 1 (IgFLNA1–15), Rod 2 (IgFLNA16–23), and C-terminal dimerization domain (IgFLNA24). Identified mutations are indicated in red. b-d Pedigrees and Sanger sequencing confirmation of identified mutations. Affected males are indicated in black squares, carrier females shown by circles with black dot. Genotyping results are shown by WT (wildtype), MT (mutant) or NT (not tested). Pedigree 1 is multiplex family with two affected half-brothers while Pedigrees 2 and 3 are of sporadic PBS cases. In all cases, co-segregation of identified mutation with PBS phenotype and maternal inheritance suggests X-linked recessive mode of inheritance
Subject 1 (Fig. 1 A1-A2) is a 29-year-old non-Hispanic white male with familial syndromic PBS (PBS + OPDSD) (RUBACE score 22). At 12 weeks gestation, he manifested bilateral severe hydronephrosis with renal dysplasia progressing to severe oligohydramnios which was treated with multiple fetal bladder aspirations. Born prematurely at 32 weeks gestation, he was initially ventilator dependent and had gastroesophageal reflux disease (GERD) requiring Nissen and gastrostomy tube. His urinary tract malformations included bilateral grade 4 VUR, urachal diverticulum, large capacity bladder, and urethral obstruction requiring genitourinary surgeries including neonatal vesicostomy, bilateral ureteral reimplantation, and urethral dilation. As an adult, he developed end stage renal disease and received a renal transplant. As he cannot urinate to completion, he empties his bladder by clean intermittent catheterization. His OPDSD features include distinct craniofacial and skeletal abnormalities including craniosynostosis with hydrocephalus requiring ventricular-atrial shunt, prominent supraorbital ridge, hypodontia, bilateral flaring of the anterior ribs, lumbar levoscoliosis, and bilateral genu valgum treated with distal femoral osteostomies (Supplementary Fig. 2: A1 and Table 1).
Subject 2 (Fig. 1 B1-B2) is a 25-year-old non-Hispanic white male with familial syndromic PBS (PBS + OPDSD) (RUBACE score 24). At 16 weeks gestation, a grossly enlarged bladder with oligohydramnios was diagnosed. Born prematurely at 32 weeks gestation, he was initially ventilator dependent for 2 months; episodes of aspiration pneumonia and gastroesophageal reflux disease (GERD) mandated Nissen and gastrostomy tube. Genitourinary anomalies included bilateral renal dysplasia necessitating neonatal temporary peritoneal dialysis, large capacity bladder and megaureters requiring neonatal vesicostomy and later partial cystectomy with bilateral tapered ureteral reimplantation, catheterizable continent vesicostomy and bilateral intraabdominal cryptorchidism. Gastrointestinal phenotypes include intestinal malrotation requiring LADD procedure and temporary jejunostomy for duodenal perforation. As an adult, he developed end stage renal disease requiring renal transplantation, which warranted a mesh abdominal wall reconstruction due to his severe degree of abdominal wall weakness at the time of transplantation. Like his half-brother, he empties his bladder by clean intermittent catheterization. His OPDSD features include prominent supraorbital ridge, down slanting palpebral fissures, proptosis, ocular hypertelorism, hypodontia, facial asymmetry, conductive hearing loss, developmental delay, cleft palate, bilateral absence of the T12 ribs, “tree-frog” feet, bilateral pars defects and spina bifida at L5, and severe scoliosis requiring multiple surgical interventions (Supplementary Fig. 2: B1-B2 and Table 1).
Subject 3 (Fig. 1 C1-C2) is a 51-year-old non-Hispanic white male from Pedigree 2 (Fig. 2c) with sporadic isolated PBS (RUBACE score 14). As a term infant, his urinary tract was devastated, presenting with bilateral ureteral obstruction with acute kidney injury requiring > 7 urinary tract reconstructions and diversions (neonatal bilateral cutaneous ureterostomies, 3 ureteral reimplantations, and ileal conduit diversion at age 6 yrs). Now, he remains with an ileal conduit, has required partial nephrectomy, and has recurrent kidney stones with renal insufficiency (CKD3, GFR 30 ml/min/1.73m2). Despite many surgeries, his abdominal wall is lax requiring recurrent hydrocele and inguinal hernia repairs after childhood orchiopexies. Phenotypic features of OPDSD in Subject 3 include mild scoliosis, a high arching palate and broad shortened distal phalanges but prominent craniofacial abnormalities are not noted (Supplementary Fig. 2: C1-C2 and Table 1).
Subject 4 (Fig. 1 D1-D2) is a 7-year-old non-Hispanic mixed race male from Pedigree 3 (Fig. 2d) with sporadic isolated PBS (RUBACE score 13). Prenatal diagnosis of obstructive uropathy with hydronephrosis who was born term but small for gestational age and did not require intubation/ventilation. Postnatally, he had bilateral pelvocaliectasis with bilateral grade 2 VUR and a large capacity bladder with urachal diverticulum. He has had urachal diverticulectomy, abdominoplasty, and bilateral orchiopexy for intraabdominal testes. Currently he has normal renal function and no VUR but empties his bladder by clean intermittent catheterization. Phenotypic features of OPDSD in Subject 4 include developmental delay, short and broadened distal phalanges, short and broad halluces, but prominent craniofacial abnormalities are not noted (Supplementary Fig. 2: D1-D2 and Table 1).
WES identifies mutations in FLNA in PBS affected patients
We first obtained DNA from the two affected maternally shared half-brothers in the multiplex family (Fig. 2b, Pedigree 1 II-1 and II-3) and performed paired end whole exome sequencing (WES) from peripheral blood DNA on both. Details on WES metrics and variant analysis are provided in Supplementary Table 1. Filtering of the variant call files for variants that met our filtering criteria (coding or impact a splice site, not reported in public databases) and shared by both half-brothers revealed FLNA c.6727C > T (GeneBank: NM_001110556.1); p.Cys2160Arg variant on chromosome X as the only recessive variant shared by both half-brothers (Supplementary Table 1). The alternative allele was identified in 100% of WES reads at this position in both affected male individuals and is not reported in ExAC or gnomAD. Sanger sequencing confirmed that both PBS half-brothers are hemizygous for this variant and their mother (Fig. 2b, Pedigree 1, I-2) is a heterozygous carrier with 95:5 ratio of skewed X-chromosome inactivation by androgen receptor methylation assay, suggesting a protective mechanism against a deleterious mutation on one X-chromosome and presumably preferred expression of the wild type allele (Supplementary Fig. 1). Although not previously clinically diagnosed, the half-brothers manifest PBS with Otopalatodigital (OPD) spectrum disorder (OPDSD) while their mother has no PBS features but has mild OPDSD phenotypic manifestations (Table 1 and Supplementary Fig. 1) [2, 44]. Although there is unconfirmed maternal family history of PBS in earlier generations (expanded pedigree in Supplementary Fig. 1), none are living to test for presence of the variant. Biallelic autosomal variants common to both brothers that met the filtering criteria were not found in this family and because a recessive or X-linked recessive mode of inheritance is hypothesized for PBS, FLNA emerged as the most likely candidate gene in this multiplex PBS kindred. The p.C2160R mutation within FLNA Rod 2 (Fig. 2A) affects a highly conserved residue of the immunoglobulin repeat 20 of FLNA (IgFLNA20), which is within the integrin interaction domain (Fig. 4a). Integrins form heterodimers (ITGα/ITGβ) that have extracellular ligand-binding loops and a C-terminal cytoplasmic interactive tail. FLNA Rod 2 binds the cytoplasmic tail of β-integrins, mediating cell contraction and remodeling of collagen matrices (in the case of FLNA/ITGβ1) [45]. In response to mechanical force, ITGβ1 recruits both FLNA and actin to ITGβ1-containing membrane focal adhesions [46]. Taken together, these data suggest that the identified PBS FLNA variant in the two half-brothers has a potential functional impact on the mechanosensing properties of FLNA through its interaction with integrin beta tails.
To further investigate whether FLNA variants cause PBS in other unrelated individuals, we searched our WES database of sporadic and familial PBS patients and found variants in FLNA in four sporadic PBS-affected individuals (Supplementary Table 2). Upon Sanger sequencing validation and inheritance testing, two of these four variants (p.Arg24Leu and p.Gly2138Cys) did not segregate with the phenotype in the family and were therefore not pursued for further investigation. Furthermore, the p.Gly2138Cys mutation did not meet our WES filtering criteria as it was reported in ExAC in three male individuals (MAF =0.00009) suggesting that this mutation is not causal for PBS.
However, the remaining two variants were in two unrelated sporadic isolated PBS male cases without OPDSD (Fig. 2c and d, Pedigree 2 II-1 and Pedigree 3 II-1). They harbor hemizygous non-synonymous, novel mutations in FLNA that fit our filtering criteria– c.4952 C > T (GeneBank: NM_001110556.1); p.Ala1448Val (Subject 3) and c.5966 G > A (GeneBank: NM_001110556.1); p.Gly2236Glu (Subject 4). Details on WES data are available in Supplementary Table 1. The variants identified in the two sporadic individuals affect highly conserved residues of FLNA and are not observed in the ExAC database. In all cases, the identified FLNA variants are maternally inherited, further supporting an X-linked recessive mode of inheritance. In summary, we identified three unreported missense point mutations across four individuals in FLNA as causal for PBS (Figs. 1 and 2).
FLNA is expressed in bladder smooth muscle
To assess the relationship between the PBS phenotype and mutant FLNA, we examined normal FLNA spatiotemporal expression. We first consulted publicly available expression databases including GenePaint and EurexpressII, transcriptome-wide compendiums of mouse in situ hybridizations during fetal life. Flna is highly expressed in the developing bladder detrusor, ureter, abdominal wall musculature, urogenital sinus mesenchyme, intestinal smooth muscle inner circular layer, discrete cardiac regions, alveolar buds of the lung as well as the sternum of the embryonic day 14.5 wild type mouse. These expression patterns correlate with the cardinal features and the extra-genitourinary manifestations of PBS [2, 3]. To compare spatial expression of FLNA in humans, we performed qPCR on RNA isolated from normal pooled adult human donor tissues and found FLNA to be highly expressed in smooth muscle-containing organs with strong enrichment in the small intestines and urinary bladder (Fig. 3a). To assess for age -related differences in expression, four pediatric normal bladder biopsies (without PBS) were evaluated by FLNA immunohistochemistry, which revealed predominant expression of FLNA within bundles of smooth muscle cells but not in intervening connective tissue (Fig. 3b). Although we did not have bladder biopsies from the four PBS males described in this report, FLNA bladder IHC on 3 other PBS cases of undiagnosed molecular cause revealed no significant change in FLNA protein immunostaining (data not shown), which is consistent with gain of function FLNA mutations that do not disturb FLNA protein quantity (see below in Discussion). FLNA expression in smooth muscle tissues, particularly the bladder detrusor, in both mice and humans is consistent with the phenotypes observed in PBS and further supports it as a novel PBS causal gene.

FLNA is expressed in human bladder smooth muscle. a qPCR across adult human tissues. FLNA expression normalized to GAPDH and relative to brain. b IHC from pediatric normal human bladder shows cytoplasmic and some nuclear smooth muscle expression of FLNA
Mutations in FLNA affect binding to integrin proteins
In order to assess the functional impact of the PBS-associated FLNA mutations, we interrogated the FLNA-integrin binding interaction, a well characterized interaction important for cell migration, polarity, and contraction [47,48,49,50]. While both ITGβ1 and 7 have been demonstrated to bind to IgFLNA21, unlike ITGβ7, ITGβ1 is highly expressed in mouse bladder and small intestinal smooth muscle as well as abdominal wall skeletal muscle tissues (GenePaint, EurexpressII) [47, 49]. Further, ITGβ1 specifically localizes to the plasma membrane of smooth muscle cells from human pediatric normal bladder tissue (Fig. 4c and d). Thus, to investigate the functional consequences of the identified FLNA mutations on the interaction between FLNA and ITGβ1, CHO (Chinese hamster ovary) cells were transfected with full length FLNA plasmids carrying the candidate PBS mutations, and cell lysates were used for pull down assays with immobilized β-integrin tails as described [43, 47, 49]. The previously studied artificial FLNA mutations I2144E (in IgFLNA20) and ΔIg20 are not PBS-related, but have been engineered to demonstrate the auto-inhibitory mechanism of IgFLNA20 on IgFLNA21 which normally blocks ITGβ1 binding to FLNA [47, 49] (Fig. 4a). ΔIg20 and I2144E exhibit enhanced FLNA binding to β-integrin tails resulting from the “open” configuration of the C/D binding face of IgFLNA21, allowing enhanced FLNA/integrin interactions without regulation from a force-induced signal [48]. These mutations were included in this study as positive controls for disruption of IgFLNA20–21 interactions. As expected, I2144E, which introduces a large polar acidic residue into IgFLNA20, strongly enhances binding to ITGβ1. Similarly, we observed a strong increase in binding of FLNA to ITGβ1 tails with the PBS FLNA C2160R mutation and to a lesser extent in the G2236E mutant when compared to WT. There was no change observed with the FLNA A1448V mutation (Fig. 4b). Comparable results were obtained in a direct binding assay using purified GST-FLNA Ig19–21 proteins (data not shown).

PBS FLNA mutations disrupt binding to integrin. a PyMol 3D protein standard cartoon of wildtype IgFLNA repeats 19 (magenta), 20 (green), and 21 (blue) showing the auto-inhibitory IgFLNA20 β-strand A on the β-strand C/D face of IgFLNA21. Shown in red are C2160 and G2236, the locations of the two FLNA residues in Ig20 and Ig21 mutated in PBS (PDB code 2J3S). The position of the IgFLNA20 β-strand A residue I2144 is shown in black; although it has not been reported mutated in humans, the I2144E mutation has been shown to lead to increased FLNA-ITGβ binding. The FLNA PBS mutation p.A1448V in Ig13 is not shown. b Pulldown assay showing that p.C2160R and p.G2236E mutations enhance binding to ITGβ1 similarly to engineered positive controls p.I2144E and p.ΔIg20. In contrast, p.A1448V binds ITGβ1 similar to WT. CHO cells were transfected with full length FLNA and bound to ITGβ1 tails on beads. c qPCR across adult human tissues shows ITGB1 expression is highest in small intestines but also strong bladder expression. ITGB1 expression normalized to GAPDH and relative to brain. d IHC from pediatric normal human bladder shows plasma membrane smooth muscle expression of ITGβ1
Discussion
In this report, we identify four surviving males (two with familial syndromic PBS + OPDSD and two with sporadic isolated PBS) to harbor three different mutations in the X-chromosome gene, FLNA, thereby expanding the phenotypic spectrum attributed to the filaminopathies. FLNA belongs to a family composed of three paralogues including Filamin A (FLNA), Filamin B (FLNB) and Filamin C (FLNC). Structurally, the filamins (FLN) form V-shaped homodimers, resulting in orthogonal crosslinking of F-actin. FLNs can be cleaved at two hinge sites into Rod1 (IgFLN1-IgFLN15) with multiple actin-binding domains (ABD) (including a high avidity F-actin binding domain in IgFLNA9–15) and Rod 2 (IgFLN16–IgFLN23) which does not bind actin but is stretch-sensing and binds many regulatory proteins [51]. In addition, each monomer of FLN also contains an N-terminal ABD and a C-terminal dimerization domain at Ig24 (Fig. 2a) [51]. The versatility of filamin functions, diversity of expression patterns and heterogeneity of binding partners is reflected in the spectrum of human disease phenotypes attributed to the filaminopathies. Deleterious mutations in FLNA, FLNB and FLNC cause malformations of the brain, cranium, face, skin, viscera, skeleton, heart, vasculature and muscle [52,53,54,55,56].
Unlike FLNB and FLNC which are autosomal genes, FLNA is a dominant X-linked gene with a wide but regionally intense expression pattern. FLNA mutations are broadly described as either loss-of-function (LOF), with reduced or absent FLNA expression, or gain-of-function (GOF), with normal expression of a full length in-frame mutant pathogenic FLNA protein [57]. FLNA mutations cause a wide spectrum of phenotypically overlapping genetic diseases [58]. The classic OPDSDs are osteochondrodysplasias including OPD1, OPD2, frontometaphyseal dysplasia (FMD), MNS, and terminal osseous dysplasia with pigmentary defects (TOD) [42]. Beyond the OPDSDs, FLNA variants have also been shown to cause XCVD, PVNH1 and PVNH4, childhood interstitial lung disease (ChILD), structural cardiac and aortic anomalies, thoracic aortic aneurysms (TAA), chronic intestinal pseudo-obstruction (CIPO), and congenital short bowel syndrome (CSBS) (Fig. 5a) [57, 59,60,61,62,63,64,65,66,67,68,69,70,71]. Typically, PVNH, XCVD, CIPO and CSBS are thought to be caused by LOF mutations while OPDSDs are caused by GOF FLNA mutations.

Human Disease Causing Mutations in FLNA in Surviving and Non-surviving Males. a FLNA disease-causing mutations cluster in ‘hot spots.’ PVNH mutations are largely found in the actin binding domain (ABD) while OPD spectrum disorders (including OPD, FMD and MNS) cluster in IgFLNA10. We identified 3 novel PBS variants: A1448V in IgFLNA13 (Rod 1), C2160R in IgFLNA20 and G2236E in IgFLNA21 (Rod 2). b Model for FLNA interaction with integrins. In a relaxed state, there is an auto-inhibitory interaction between Ig20–21 that masks the integrin binding site on Ig21. Normally, mechanical stretching can cause exposure of the integrin binding site on Ig21. Mutations in Ig19–21 result in loss of FLNA function as a stretch sensor and disruption of the auto-inhibitory interaction between Ig20–21 causing constitutive exposure of the integrin binding site on Ig21 and thereby enhanced binding to integrins
FLNA mutations in males is often lethal, however, a small subset with missense FLNA mutations have been reported to survive beyond the first weeks of postnatal life. (Fig. 5a) [41, 67, 72]. Exceedingly rarer are adult surviving males with FLNA mutations. Herein, we document three additional surviving adults with FLNA mutations and draw focus upon the genitourinary phenotypes caused by FLNA mutation. Past publications have broadly noted “obstructive uropathy” or “genitourinary abnormalities” in rare male patients who died neonatally with FLNA mutations. In 1987, prune belly sequence was observed in a MNS patient not sequenced for FLNA [73]. More recently, four males with lethal MNS and FLNA exon 22 mutations disrupting Ig10 have been described with genitourinary abnormalities including omphalocele, megacystis, and/or “prune belly-like” phenotype of abdominal wall laxity [58, 74, 75]. A lethal form of FMD from FLNA exon 22 mutation was noted in a male with distended abdomen, megaureters and hydronephrosis [76]. Additionally, a 2009 report of an Xq28 duplication (which includes FLNA) described a kindred with several males who died as infants, one of which was diagnosed with prune belly syndrome and another with a grossly distended bladder after birth [77]. Overall, there is not a clear genotype-phenotype correlation when these old published cases are added to our series, other than the fact that most MNS have FLNA exon 22 mutations within Ig10. As FLNA Ig10 is quite remote from the FLNA Ig19–21 domain, it is biologically unclear how missense mutations in Ig10 yield the obstructive uropathy phenotype. As these genotypic differences exist and since PBS due to deleterious FLNA mutations is seen with or without OPDSD, we prefer to segregate our PBS phenotype from the MNS phenotype, as depicted in Fig. 5a.
FLNA is a critical intracellular connecting protein between the cytoskeleton and transmembrane complexes that bind to extracellular matrix. At the protein level, the FLNA:integrin interaction is highly regulated, as in vitro excessive integrin binding of FLNA prevents efficient actin remodeling and cell motility [78]. Specifically, the FLNA repeat domains Ig20 and Ig21 are critical for this regulation, functioning as a stretch mechanosensor [79, 80]. The binding site for β-integrin tails is on IgFLNA21 but when FLNA is not stretched, this site is blocked by the β-strand A of IgFLNA20, suggesting an auto-inhibitory mechanism regulating FLNA:integrin binding [48]. A mechanism for up-regulation of FLNA integrin binding is force-induced mechanical stretching of FLNA, permitting integrin binding. When FLNA is stretched, IgFLNA20 is disengaged from IgFLNA21 thereby exposing the IgFLNA21 C/D face for binding of integrins to IgFLNA21 and stabilizing the stretched FLNA conformation. Once force is released, the IgFLNA20 B-strand refolds back onto IgFLNA21, displacing integrin [45, 46, 81,82,83,84,85]. Two of our detected PBS IgFLNA19–21 variants alter binding of FLNA to β-integrin tails. The PBS mutations C2160R (in IgFLNA20) and G2236E (in IgFLNA21) are both substitutions of small uncharged residues to large charged residues with multiple side chains. The newly introduced, large charged residues likely significantly contribute to the disruption of the IgFLNA20–21 interface, presumably stabilizing the exposure of the integrin binding site on IgFLNA21 leading to enhanced binding of ITGβ1 tails similarly to the engineered I2144E and ΔIg20 mutations (Fig. 5b). Altered transmembrane receptor binding to FLNA and ligand-independent phosphorylation of FLNA has been reported in the IgFLNA20 P2204L mutation found in FMD [86]. Improper mechanosensing properties of FLNA likely leads to apoptosis in force-loaded cells [87].
The Rod1 A1448V variant is located in the N-terminal region of Ig13, which has not been reported to bind integrins. Unlike the ligand binding IgFLNA19 and IgFLNA21 Class A repeats, Ig13 is a member of the class D Ig repeats (which includes IgFLNA10). IgFLNA10 has been crystalized and functional implications of mutations causal for MNS have been assessed such that the p.A1188T and p.S1199L MNS mutations are predicted to alter hydrophobic packing of IgFLNA10 and possibly impact FLNA stability [88]. Together, these data suggest that C2160R and G2236E may indeed cause disease via disrupted integrin-dependent function of FLNA while the A1448V substitution may affect an integrin-independent function of FLNA.
FLNA is critical in embryonic development. 100% of hemizygous male mice null for Flna die by E14.5 with widespread hemorrhage, incomplete heart outflow tract septation (common arterial trunk), ventricular septal defects, and mitral valve dysplasia [89, 90]. Coarse dilated blood vessels within many tissues led to organ malformations and angiogenesis was disrupted by aberrant adherens junctions in endothelial cells [89]. Additional midline malformations in these male mice include non-fused split sternum, umbilical hernia and cleft palate [90]. Flna binds actin-nucleating Formin 2 (Fmn2) and Flna + Fmn2 null mice exhibit microcephaly, thoracoabdominal schisis, thinned ventral body wall (muscle, ribs, and sternum) and shortened gut lengths when compared to WT embryos, partially due to fewer proliferating mesenchymal cells in the sternum and ribcage [91]. The inbred Long Evans orl rat has spontaneous cryptorchidism secondary to abnormal formation of the gubernaculum, the cord-like ventral abdominal wall appendage that normally drags the testis into the scrotum during embryonic life. The orl rat has altered Flna expression in its gubernaculum, which is comprised of a core of mesenchymal cells with associated extracellular matrix and localized striated muscle [92]. These data stress the essential role of Flna for normal cardiac, vascular, skeletal, abdominal wall, gubernaculum, and gastrointestinal development during embryogenesis. Beyond embryonic life, conditional deletion of Flna in adult murine smooth muscle lineages such as vascular smooth muscle cells (VSMCs) induces hypertrophic remodeling of the carotid artery and aorta [93]. Zhu et al observed that when rabbit aortic VSMCs are overproliferating or actively migrating, FLN is expressed at elevated levels. Conversely, when expression of FLN is decreased, VSMCs return to the “contractile” phenotype, suggesting a key role in VSMCs phenotype switching [94]. In cultured cells, Flna has been shown to interact with Mkl1 and promote a Srf-dependent smooth muscle transcriptional program [95].
Our PBS cases highlight the role of FLNA in muscle development and function. In humans, several FLNA disease phenotypes manifest smooth muscle dysfunction (leiomyopathy), including TAA, CIPO and CSBS. Recent work by Jenkins et al. has explored why some males with 5′ mutations phenotypically only manifest CIPO. They identified tissue-specific differential expression of two FLNA transcripts and suggest the longer protein isoform (ATG+ 1) is crucial for smooth muscle development [57]. Alternatively, whether there is a FLNA mutational hotspot for leiomyopathies is not clear. In support of this concept, a familial case of CIPO and intestinal malrotation in two affected brothers was shown to be caused by a 4 bp deletion of FLNA exon 40 causing an in-frame exon skipping affecting FLNAIg20 [67]. Very few mutations have been identified in the highly studied IgFLNA19–21 region, however, this triplicate domain of FLNA is not only important for binding of many interacting partners including integrins, but potentially for FLNA function unique to smooth muscle cells and now PBS [39, 40, 64, 96, 97].
The three cardinal features of PBS can thus be explained by deranged FLNA signaling. However, PBS is a complex phenotype, ranging from neonatal lethality to mild manifestations. PBS lethality is most commonly associated with in utero oligohydramnios from low volume of fetal urine expelled from the urinary system. This low urine volume may be due to poor urine production by a malformed fetal kidney and/or improper bladder and ureteral emptying. Oligohydramnios is also associated with abnormal lung development (bronchopulmonary dysplasia) often associated with respiratory death. In mice, Flna is expressed in the fetal alveoli and airway smooth muscle cells, fetal glomeruli and urinary tract urothelium and smooth muscle cells – all of which are crucial for postnatal life.
At this point, many cases of PBS remain genetically undefined. Other plausible genes implicated in the pathogenesis of PBS include variants in CHRM3 (the muscarinic cholinergic receptor M3 responsible for bladder smooth muscle contraction), ACTA2 and ACTG2 (two actin genes expressed in smooth muscle), HNF1β (the embryonic transcription factor hepatocyte nuclear factor 1β) and STIM1 (the stromal interaction molecule 1 which has calcium channel regulatory activity). Variants in these genes only explain single cases or one independent consanguineous family [32,33,34,35,36, 98, 99]. Our WES data has not identified exonic mutations in CHRM3, HNF1β or STIM1 in PBS patients, although we have found one individual with a previously unreported heterozygous variant in ACTA2 and two individuals with previously unreported heterozygous mutations in ACTG2. Whether the reported actin mutations directly interfere with FLNA binding is unknown. The long term implications of making a molecular diagnosis in cases of PBS are significant, as PBS patients harboring a FLNA or ACTA2 mutation may need to adopt surveillance strategies for cardiovascular disease such as moya-moya or TAA [69, 71, 100]. Overall, this suggests that PBS is not monogenic, but may be caused by mutations in multiple genes and that other causal genes remain to be identified [32].
Our study does have limitations. Our PBS study did not include FLNA WES in perinatal PBS deceased cases. Our cohort of surviving patients with PBS likely induces a survivorship bias, thus limiting and underestimating the number of PBS cases (lethal or living) with FLNA mutations. We did not demonstrate any recurrent FLNA mutations in PBS in our small cohort. Lastly, our study suffers from the lack of additional tissues or patient derived cells for further correlative testing and the lack of a PBS mouse model. Nevertheless, our data is compelling.
Conclusions
In summary, we have identified three hemizygous mutations in FLNA in one multiplex kindred and two sporadic PBS males with or without OPDSD phenotypes who have survived even to adulthood. This report highlights the role of FLNA in rhabdomyopathy, cardiomyopathy and leiomyopathy. It also represents the first proposed PBS candidate gene to support an X-linked recessive mode of inheritance and the first candidate gene identified in both familial and unrelated sporadic individuals, representing the largest number of cases with mutations in the same gene as causal for the congenital myopathy PBS.
Availability of data and materials
The datasets generated during the current study are available in Clinvar under the following accession numbers: VCV000801015, VCV000800569, VCV000800568.
Abbreviations
- ACTA2:
-
Actin, alpha 2, smooth muscle, aorta
- ACTG2:
-
Actin, gamma 2, smooth muscle, enteric
- ADHD:
-
Attention-deficit/hyperactivity disorder
- chILD:
-
Chronic interstitial lung disease
- CHRM3:
-
Cholinergic receptor muscarinic 3
- CIPO:
-
Chronic intestinal pseudoobstruction
- CKD:
-
Chronic kidney disease
- CSBS:
-
Chronic short bowel syndrome
- FLNA:
-
Filamin A
- FLNB:
-
Filamin B
- FLNC:
-
Filamin C
- FMD:
-
Frontotmetaphyseal dysplasia
- GERD:
-
Gastroesophageal reflux disease
- GOF:
-
Gain of function
- HNF1b:
-
Hepatocyte nuclear factor 1 β
- Ig:
-
Immunoglobulin repeat
- ITGB1:
-
Integrin β 1
- ITGα:
-
Integrin alpha
- LOF:
-
Loss of function
- MAF:
-
Minor allele frequency
- MNS:
-
Melnick-Needles syndrome
- OPD1/2:
-
Ototopalatodigital syndrome type 1/2
- OPDSD:
-
Otopalatodigital spectrum disorders
- PBS:
-
Prune Belly Syndrome
- PVNH:
-
Periventricular nodular heterotopia
- RUBACE:
-
Renal, ureter, bladder/outlet, abdominal wall, cryptorchidism, extra-genitourinary
- S/P:
-
Status post
- STIM1:
-
Stromal interaction molecule 1 precursor
- TAA:
-
Thoracic aortic aneurysm
- VUR:
-
Vesicoureteral reflux
- WES:
-
Whole exome sequencing
- XCVD:
-
X-linked cardiac valvular dystrophy
References
Routh JC, Huang L, Retik AB, Nelson CP. Contemporary epidemiology and characterization of newborn males with prune belly syndrome. Urology. 2010;76(1):44–8.
Wong DG, Arevalo MK, Passoni NM, Iqbal NS, Jascur T, Kern AJ, et al. Phenotypic severity scoring system and categorisation for prune belly syndrome: application to a pilot cohort of 50 living patients. BJU Int. 2018.
Grimsby GM, Harrison SM, Granberg CF, Bernstein IH, Baker LA. Impact and frequency of extra-genitourinary manifestations of prune belly syndrome. J Pediatr Urol. 2015;11(5):280 e1–6.
Druschel CM. A descriptive study of prune belly in New York state, 1983 to 1989. Arch Pediatr Adolesc Med. 1995;149(1):70–6.
Lopes RI, Tavares A, Srougi M, Denes FT. 27 years of experience with the comprehensive surgical treatment of prune belly syndrome. J Pediatr Urol. 2015;11(5):276 e1–7.
Musone D, Nicosia V, D'Alessandro R, Treglia A, Saltarelli G, Montella M, et al. [Peritoneal dialysis in adult patients with prune belly syndrome: an impossible challenge?]. G Ital Nefrol. 2013;30(4).
Yalcinkaya F, Bonthuis M, Erdogan BD, van Stralen KJ, Baiko S, Chehade H, et al. Outcomes of renal replacement therapy in boys with prune belly syndrome: findings from the ESPN/ERA-EDTA registry. Pediatr Nephrol. 2018;33(1):117–24.
Becknell B, Pais P, Onimoe G, Rangarajan H, Schwaderer AL, McHugh K, et al. Hepatoblastoma and prune belly syndrome: a potential association. Pediatr Nephrol. 2011;26(8):1269–73.
Wheatley JM, Stephens FD, Hutson JM. Prune-belly syndrome: ongoing controversies regarding pathogenesis and management. Semin Pediatr Surg. 1996;5(2):95–106.
Stephens FD, Gupta D. Pathogenesis of the prune belly syndrome. J Urol. 1994;152(6 Pt 2):2328–31.
Nivelon-Chevallier A, Feldman JP, Justrabo E, Turc-Carel C. Trisomy 18 and prune belly syndrome. J Genet Hum. 1985;33(5):469–74.
Amacker EA, Grass FS, Hickey DE, Hisley JC. An association of prune belly anomaly with trisomy 21. Am J Med Genet. 1986;23(4):919–23.
Beckmann H, Rehder H, Rauskolb R. Prune belly sequence associated with trisomy 13. Am J Med Genet. 1984;19(3):603–4.
Frydman M, Magenis RE, Mohandas TK, Kaback MM. Chromosome abnormalities in infants with prune belly anomaly: association with trisomy 18. Am J Med Genet. 1983;15(1):145–8.
Hoagland MH, Frank KA, Hutchins GM. Prune-belly syndrome with prostatic hypoplasia, bladder wall rupture, and massive ascites in a fetus with trisomy 18. Arch Pathol Lab Med. 1988;112(11):1126–8.
McKeown CM, Donnai D. Prune belly in trisomy 13. Prenat Diagn. 1986;6(5):379–81.
Metwalley KA, Farghalley HS, Abd-Elsayed AA. Prune belly syndrome in an Egyptian infant with Down syndrome: a case report. J Med Case Rep. 2008;2:322.
Guillen DR, Lowichik A, Schneider NR, Cohen DS, Garcia S, Zinn AR. Prune-belly syndrome and other anomalies in a stillborn fetus with a ring X chromosome lacking XIST. Am J Med Genet. 1997;70(1):32–6.
Ramasamy R, Haviland M, Woodard JR, JG B. Patterns of inheritance in familial prune belly syndrome. Urology. 2005;65(6):1227.
Garlinger P. J O. prune belly syndrome: possible genetic implications. Birth Defects. 1974;10:173–80.
Balaji KC, Patil A, Townes PL, Primack W, Skare J, Hopkins T. Concordant prune belly syndrome in monozygotic twins. Urology. 2000;55(6):949.
Petersen DS, Fish L, Cass AS. Twins with congenital deficiency of abdominal musculature. J Urol. 1972;107(4):670–2.
Grenet P, Le Calve G, Badoual J. al e. congenital aplasia of the abdominal wall: a familial case. Ann Pediatr. 1972;19:523–8.
Harley LM, Chen Y, WH R. Prune belly syndrome. J Urol. 1972;108:174–6.
Afifi AK. Rebeiz J, mire J, al e. the myopathology of the prune belly syndrome. J Neurol Sci. 1972;15:153–65.
Riccardi VM, CM G. The prune belly anomaly: heterogeneity and superficial X-linkage mimicry. J Med Genet. 1977;14:266–70.
Lockhart JL, Reeve HR, Bredael JJ, Krueger RP. Siblings with prune belly syndrome and associated pulmonic stenosis, mental retardation, and deafness. Urology. 1979;14(2):140–2.
Gaboardi F, Sterpa A, Thiebat E. al e. prune-belly syndrome: report of three siblings. Helv Paediatr Acta. 1982;37:283–8.
Adeyokunnu AA, JB F. Prune belly syndrome in two siblings and a first cousin: possible genetic implications. Am J Dis Child. 1982;136:23–5.
Feige A, Fiedler K, Rempen A. al e. prenatal diagnosis of prune belly syndrome occurring in siblings in 2 consecutive pregnancies. Z Geburtshilfe Perinatol. 1984;188:239–43.
Chan Y, L B. Vertically transmitted hypoplasia of the abdominal wall musculature. Clin Dysmorphol. 2004;13:7–10.
Granberg CF, Harrison SM, Dajusta D, Zhang S, Hajarnis S, Igarashi P, et al. Genetic basis of prune belly syndrome: screening for HNF1beta gene. J Urol. 2012;187(1):272–8.
Richer J, Milewicz DM, Gow R, de Nanassy J, Maharajh G, Miller E, et al. R179H mutation in ACTA2 expanding the phenotype to include prune-belly sequence and skin manifestations. Am J Med Genet A. 2012;158A(3):664–8.
Wangler MF, Gonzaga-Jauregui C, Gambin T, Penney S, Moss T, Chopra A, et al. Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome. PLoS Genet. 2014;10(3):e1004258.
Brodsky MC, Turan KE, Khanna CL, Patton A, Kirmani S. Congenital mydriasis and prune belly syndrome in a child with an ACTA2 mutation. J AAPOS. 2014;18(4):393–5.
Weber S, Thiele H, Mir S, Toliat MR, Sozeri B, Reutter H, et al. Muscarinic acetylcholine receptor M3 mutation causes urinary bladder disease and a prune-belly-like syndrome. Am J Hum Genet. 2011;89(5):668–74.
Boghossian NS, Sicko RJ, Giannakou A, Dimopoulos A, Caggana M, Tsai MY, et al. Rare copy number variants identified in prune belly syndrome. Eur J Med Genet. 2017.
Iqbal NS, Jascur TA, Harrison S, Chen C, Arevalo MK, Wong D, et al. Copy number variations in a population with prune belly syndrome. Am J Med Genet A. 2018;176(11):2276–83.
Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2(2):138–45.
Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6(11):1034–8.
Cannaerts E, Shukla A, Hasanhodzic M, Alaerts M, Schepers D, Van Laer L, et al. FLNA mutations in surviving males presenting with connective tissue findings: two new case reports and review of the literature. BMC Med Genet. 2018;19(1):140.
Robertson S. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, LJH B, et al., editors. Otopalatodigital Spectrum Disorders. Seattle (WA): GeneReviews(R); 1993.
Lad Y, Harburger DS, Calderwood DA. Integrin cytoskeletal interactions. Methods Enzymol. 2007;426:69–84.
Moutton S, Fergelot P, Naudion S, Cordier MP, Sole G, Guerineau E, et al. Otopalatodigital spectrum disorders: refinement of the phenotypic and mutational spectrum. J Hum Genet. 2016;61(8):693–9.
Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, et al. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20(14):3224–38.
Glogauer M, Arora P, Chou D, Janmey PA, Downey GP, McCulloch CA. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J Biol Chem. 1998;273(3):1689–98.
Calderwood DA, Huttenlocher A, Kiosses WB, Rose DM, Woodside DG, Schwartz MA, et al. Increased filamin binding to beta-integrin cytoplasmic domains inhibits cell migration. Nat Cell Biol. 2001;3(12):1060–8.
Lad Y, Kiema T, Jiang P, Pentikainen OT, Coles CH, Campbell ID, et al. Structure of three tandem filamin domains reveals auto-inhibition of ligand binding. EMBO J. 2007;26(17):3993–4004.
Pfaff M, Liu S, Erle DJ, Ginsberg MH. Integrin beta cytoplasmic domains differentially bind to cytoskeletal proteins. J Biol Chem. 1998;273(11):6104–9.
Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, Wegener KL, et al. The molecular basis of filamin binding to integrins and competition with Talin. Mol Cell. 2006;21(3):337–47.
Nakamura F, Osborn TM, Hartemink CA, Hartwig JH, Stossel TP. Structural basis of filamin a functions. J Cell Biol. 2007;179(5):1011–25.
Lu J, Lian G, Lenkinski R, De Grand A, Vaid RR, Bryce T, et al. Filamin B mutations cause chondrocyte defects in skeletal development. Hum Mol Genet. 2007;16(14):1661–75.
Robertson S. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, et al., editors. FLNB-Related Disorders. Seattle (WA): GeneReviews((R)); 1993.
Duff RM, Tay V, Hackman P, Ravenscroft G, McLean C, Kennedy P, et al. Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am J Hum Genet. 2011;88(6):729–40.
Gemelli C, Prada V, Fiorillo C, Fabbri S, Maggi L, Geroldi A, et al. A novel mutation in the N-terminal acting-binding domain of Filamin C protein causing a distal myofibrillar myopathy. J Neurol Sci. 2019;398:75–8.
Vorgerd M, van der Ven PF, Bruchertseifer V, Lowe T, Kley RA, Schroder R, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77(2):297–304.
Jenkins ZA, Macharg A, Chang CY, van Kogelenberg M, Morgan T, Frentz S, et al. Differential regulation of two FLNA transcripts explains some of the phenotypic heterogeneity in the loss-of-function filaminopathies. Hum Mutat. 2018;39(1):103–13.
Naudion S, Moutton S, Coupry I, Sole G, Deforges J, Guerineau E, et al. Fetal phenotypes in otopalatodigital spectrum disorders. Clin Genet. 2016;89(3):371–7.
Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin a cause diverse malformations in humans. Nat Genet. 2003;33(4):487–91.
Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21(6):1315–25.
Sheen VL, Dixon PH, Fox JW, Hong SE, Kinton L, Sisodiya SM, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10(17):1775–83.
Moro F, Carrozzo R, Veggiotti P, Tortorella G, Toniolo D, Volzone A, et al. Familial periventricular heterotopia: missense and distal truncating mutations of the FLN1 gene. Neurology. 2002;58(6):916–21.
Kakita A, Hayashi S, Moro F, Guerrini R, Ozawa T, Ono K, et al. Bilateral periventricular nodular heterotopia due to filamin 1 gene mutation: widespread glomeruloid microvascular anomaly and dysplastic cytoarchitecture in the cerebral cortex. Acta Neuropathol. 2002;104(6):649–57.
Robertson SP. Filamin a: phenotypic diversity. Curr Opin Genet Dev. 2005;15(3):301–7.
Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, et al. Mutations in the gene encoding filamin a as a cause for familial cardiac valvular dystrophy. Circulation. 2007;115(1):40–9.
van der Werf CS, Halim D, Verheij JB, Alves MM, Hofstra RM. Congenital short bowel syndrome: from clinical and genetic diagnosis to the molecular mechanisms involved in intestinal elongation. Biochim Biophys Acta. 2015;1852(11):2352–61.
Oda H, Sato T, Kunishima S, Nakagawa K, Izawa K, Hiejima E, et al. Exon skipping causes atypical phenotypes associated with a loss-of-function mutation in FLNA by restoring its protein function. Eur J Hum Genet. 2016;24(3):408–14.
Sasaki E, Byrne AT, Phelan E, Cox DW, Reardon W. A review of filamin A mutations and associated interstitial lung disease. Eur J Pediatr. 2018.
Chen MH, Choudhury S, Hirata M, Khalsa S, Chang B, Walsh CA. Thoracic aortic aneurysm in patients with loss of function Filamin a mutations: clinical characterization, genetics, and recommendations. Am J Med Genet A. 2018;176(2):337–50.
Karimi A, Milewicz DM. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can J Cardiol. 2016;32(1):26–34.
Morisaki T, Morisaki H. Genetics of hereditary large vessel diseases. J Hum Genet. 2016;61(1):21–6.
Walsh M, Hebbard G, Trainer A. Update on a previously reported male with a FLNA missense mutation. European journal of human genetics : EJHG. 2017.
Donnenfeld AE, Conard KA, Roberts NS, Borns PF, Zackai EH. Melnick-needles syndrome in males: a lethal multiple congenital anomalies syndrome. Am J Med Genet. 1987;27(1):159–73.
Santos HH, Garcia PP, Pereira L, Leao LL, Aguiar RA, Lana AM, et al. Mutational analysis of two boys with the severe perinatally lethal Melnick-needles syndrome. Am J Med Genet A. 2010;152A(3):726–31.
Spencer C, Lombaard H, Wise A, Krause A, Robertson SP. A recurrent mutation causing Melnick-needles syndrome in females confers a severe, lethal phenotype in males. Am J Med Genet A. 2018;176(4):980–4.
Giuliano F, Collignon P, Paquis-Flucklinger V, Bardot J, Philip N. A new three-generational family with frontometaphyseal dysplasia, male-to-female transmission, and a previously reported FLNA mutation. Am J Med Genet A. 2005;132A(2):222.
Clayton-Smith J, Walters S, Hobson E, Burkitt-Wright E, Smith R, Toutain A, et al. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur J Human Genet. 2009;17(4):434–43.
Kim H, Sengupta A, Glogauer M, McCulloch CA. Filamin a regulates cell spreading and survival via beta1 integrins. Exp Cell Res. 2008;314(4):834–46.
Chen H, Chandrasekar S, Sheetz MP, Stossel TP, Nakamura F, Yan J. Mechanical perturbation of filamin a immunoglobulin repeats 20-21 reveals potential non-equilibrium mechanochemical partner binding function. Sci Rep. 2013;3:1642.
Rognoni L, Most T, Zoldak G, Rief M. Force-dependent isomerization kinetics of a highly conserved proline switch modulates the mechanosensing region of filamin. Proc Natl Acad Sci U S A. 2014;111(15):5568–73.
Chen HS, Kolahi KS, Mofrad MR. Phosphorylation facilitates the integrin binding of filamin under force. Biophys J. 2009;97(12):3095–104.
D'Addario M, Arora PD, Ellen RP, McCulloch CA. Interaction of p38 and Sp1 in a mechanical force-induced, beta 1 integrin-mediated transcriptional circuit that regulates the actin-binding protein filamin-a. J Biol Chem. 2002;277(49):47541–50.
Johnson CP, Tang HY, Carag C, Speicher DW, Discher DE. Forced unfolding of proteins within cells. Science. 2007;317(5838):663–6.
Kamm RD, Kaazempur-Mofrad MR. On the molecular basis for mechanotransduction. Mechanics Chem Biosyst. 2004;1(3):201–9.
Pentikainen U, Ylanne J. The regulation mechanism for the auto-inhibition of binding of human filamin a to integrin. J Mol Biol. 2009;393(3):644–57.
Ithychanda SS, Dou K, Robertson SP, Qin J. Structural and thermodynamic basis of a frontometaphyseal dysplasia mutation in filamin a. J Biol Chem. 2017;292(20):8390–400.
Pinto VI, Senini VW, Wang Y, Kazembe MP, McCulloch CA. Filamin a protects cells against force-induced apoptosis by stabilizing Talin- and vinculin-containing cell adhesions. FASEB J. 2014;28(1):453–63.
Page RC, Clark JG, Misra S. Structure of filamin a immunoglobulin-like repeat 10 from Homo sapiens. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67(Pt 8):871–6.
Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, et al. Filamin a (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci U S A. 2006;103(52):19836–41.
Hart AW, Morgan JE, Schneider J, West K, McKie L, Bhattacharya S, et al. Cardiac malformations and midline skeletal defects in mice lacking filamin a. Hum Mol Genet. 2006;15(16):2457–67.
Lian G, Kanaujia S, Wong T, Sheen V. FilaminA and Formin2 regulate skeletal, muscular, and intestinal formation through mesenchymal progenitor proliferation. PLoS One. 2017;12(12):e0189285.
Barthold JS, McCahan SM, Singh AV, Knudsen TB, Si X, Campion L, et al. Altered expression of muscle- and cytoskeleton-related genes in a rat strain with inherited cryptorchidism. J Androl. 2008;29(3):352–66.
Retailleau K, Arhatte M, Demolombe S, Jodar M, Baudrie V, Offermanns S, et al. Smooth muscle filamin a is a major determinant of conduit artery structure and function at the adult stage. Pflugers Arch. 2016;468(7):1151–60.
Zhu G, Chen H, Zhang W. Phenotype switch of vascular smooth muscle cells after siRNA silencing of filamin. Cell Biochem Biophys. 2011;61(1):47–52.
Kircher P, Hermanns C, Nossek M, Drexler MK, Grosse R, Fischer M, et al. Filamin A interacts with the coactivator MKL1 to promote the activity of the transcription factor SRF and cell migration. Sci Signal. 2015;8(402):ra112.
Ohta Y, Hartwig JH, Stossel TP. FilGAP, a rho- and ROCK-regulated GAP for Rac binds filamin a to control actin remodelling. Nat Cell Biol. 2006;8(8):803–14.
Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adhes Migr. 2011;5(2):160–9.
Haeri S, Devers PL, Kaiser-Rogers KA, Moylan VJ Jr, Torchia BS, Horton AL, et al. Deletion of hepatocyte nuclear factor-1-beta in an infant with prune belly syndrome. Am J Perinatol. 2010;27(7):559–63.
Murray PJ, Thomas K, Mulgrew CJ, Ellard S, Edghill EL, Bingham C. Whole gene deletion of the hepatocyte nuclear factor-1b gene in a patient with the prune-belly syndrome. Nephrol Dial Transplant. 2008;23:2412–5.
Ziganshin BA, Bailey AE, Coons C, Dykas D, Charilaou P, Tanriverdi LH, et al. Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg. 2015;100(5):1604–11.
Acknowledgements
The authors warmly thank the wonderful families who contributed and express deepest gratitude to the Prune Belly Syndrome Network (www.prunebelly.org) for their invaluable help. L.B. is supported by a grant from the National Institutes of Health (DK100483) and the Seay Endowment for Pediatric Urologic Research. N.I. is supported by an American Urology Association Urology Care Foundation Research Scholars Award. We kindly thank the Calderwood laboratory (Yale University) for plasmids and ITGB1/7 tails. We are grateful to the UT Southwestern McDermott Sequencing Center (WES analysis) and also acknowledge the assistance of the University of Texas Southwestern Tissue Resource, a shared resource at the Simmons Comprehensive Cancer Center supported in part by the National Cancer Institute under award number 5P30CA142543.
Funding
L.B is supported by a grant from the National Institutes of Health (DK100483) and the Seay Endowment which provided funding for all sequencing, recruitment and functional experiments. N.I. is supported by an American Urology Association Urology Care Foundation Research Scholars Award which provided salary support for carrying out work for the project. The funding bodies played no role in the design of the study and collection, analysis and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
NSI made substantial contributions to the design of the work, analyzed and interpreted the patient WES data, performed expression analyses and drafted the manuscript. TAJ made substantial contributions to the design of the work, analyzed and interpreted the patient WES data and performed binding experiments. SMH made substantial contributions to the design of the work, analyzed and interpreted all data and was a major contributor in writing the manuscript. ABE made significant contributions to interpretation of patient clinical data and generated summary phenotyping table. LTS assisted in interpretation of crystal structure data. ESC substantially contributed to generation of all plasmids and constructs for functional experiments. MKA contributed to patient phenotypic analysis and RUBACE scoring. CC substantially contributed to interpretation of patient clinical data. SZ contributed to sequencing validation and assisted in all assays. AJK contributed to interpretation of patient clinical data and RUBACE scoring. AES reviewed patient medical photographs to interpret phenotype. EJS was instrumental to patient recruitment and gathering all patient data. CX processed and analyzed all WES data. LAB was instrumental to the study design, interpretation of all data and writing of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all individuals included in the study which was approved by the institutional review board at UT Southwestern Medical Center and all procedures followed were in accordance with the ethical standards of the relevant committees on human experimentation. In the case of children or those cognitively impaired and unable, written informed consent was obtained from the parent/legal guardian.
Consent for publication
All study participants consented to publication of photographs, clinical and genetic data which was approved by the institutional review board at UT Southwestern Medical Center.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Whole exome sequencing metrics and variant calling. Figure S1. X-linked recessive mode of inheritance hypothesized in pedigree 2. Table S2. List of identified FLNA variants in PBS cohort. Figure S2. Images of PBS affected subjects hands and feet.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Iqbal, N.S., Jascur, T.A., Harrison, S.M. et al. Prune belly syndrome in surviving males can be caused by Hemizygous missense mutations in the X-linked Filamin A gene. BMC Med Genet 21, 38 (2020). https://doi.org/10.1186/s12881-020-0973-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-0973-x