
Abstract
Background
Dilated cardiomyopathy (DCM) is a condition characterized by dilatation and systolic dysfunction of the left ventricle in the absence of severe coronary artery disease or abnormal loading conditions. Mutations in the titin (TTN) and lamin A/C (LMNA) genes are the two most significant contributors in familial DCM. Previously mutations in the desmoplakin (DSP) gene have been associated with arrhythmogenic right ventricular cardiomyopathy (ARVC) and more recently with DCM.
Methods
We describe the cardiac phenotype related to a DSP mutation which was identified in ten unrelated Finnish index patients using next-generation sequencing. Sanger sequencing was used to verify the presence of this DSP variant in the probands’ relatives. Medical records were obtained, and clinical evaluation was performed.
Results
We identified DSP c.6310delA, p.(Thr2104Glnfs*12) variant in 17 individuals of which 11 (65%) fulfilled the DCM diagnostic criteria. This pathogenic variant presented with left ventricular dilatation, dysfunction and major ventricular arrhythmias. Two patients showed late gadolinium enhancement (LGE) and myocardial edema on cardiac magnetic resonance imaging (MRI) that may suggest inflammatory process at myocardium.
Conclusions
The patients diagnosed with DCM showed an arrhythmogenic phenotype as well as SCD at young age supporting the recently proposed concept of arrhythmogenic cardiomyopathy. This study also demonstrates relatively low penetrance of truncating DSP variant in the probands’ family members by the age of 40. Further studies are needed to elucidate the possible relations between myocardial inflammation and pathogenic DSP variants.
Similar content being viewed by others


Background
Dilated cardiomyopathy (DCM) is characterized by left ventricular systolic dysfunction and dilatation of left ventricle in the absence of severe coronary artery disease or abnormal loading conditions such as hypertension or valvular disease [1]. Diagnosis is traditionally made by cardiac imaging, usually echocardiography. DCM is typically an adult-onset disease, but disease onset may take place already in infancy [2]. There is a wide variability in phenotypic expression and severity as clinical presentation varies from asymptomatic to end-stage heart failure or even sudden cardiac death (SCD) [2]. The prevalence of DCM in the general population is still unknown and it has previously been estimated to be 1:2500–1:3000 [3, 4]. These figures, however, are based on old data and the prevalence is now estimated to be much higher [5].
Familial DCM is typically considered to be a monogenic disorder and most commonly autosomal dominant inheritance has been reported [1, 2, 6, 7]. However, X-linked, recessive and mitochondrial inheritance patterns have been observed [6]. As much as 30–50% of DCM is thought to be genetic or familial [6, 8]. Over 40 genes have been related to DCM [8] and the most common genetic cause is a truncating variant in titin gene (TTN) [6, 9,10,11]. Mutations of lamin A/C gene (LMNA) are the second most common cause of DCM, causing 5–8% of familial DCM [12]. The most common causative genes in order of prevalence are TTN, LMNA, DSP, MYH7 (beta-myosin heavy chain), RBM20 (RNA binding motif protein 20), TNNT2 (Troponin 2), TPM1 (tropomyosin 1), FLNC (filamin C) and DES (desmin) [13, 14]. Mutations in sarcomeric proteins have some overlap to HCM [6]. Various genes coding cytoskeleton proteins such as DSC2 (desmocollin-2) and DSG2 (desmoglein 2) have also been associated with DCM and arrhythmogenic right ventricular cardiomyopathy (ARVC) [6].
DSP codes for the protein desmoplakin, which binds intermediate filament proteins to desmosomal plaques and is thus an essential part of a functional desmosome [15, 16]. Desmosomes are intercellular junctions that provide mechanical strength to tissues undergoing physical stress [15].
Earlier mutations in the DSP gene have mostly been associated with ARVC, which is an inherited cardiac disorder affecting usually the right ventricle [17,18,19]. The disorder is characterized by fibrofatty replacement, abnormal contraction and dilatation of the right ventricle (RV) [17, 20]. Ventricular arrhythmias often occur and may even lead to sudden cardiac death, especially in young people and athletes [20]. The diagnosis is based on the rather complex revised 2010 Task Force Criteria, and is a combination of major and minor criteria from different areas including RV function and structure, electrocardiogram (ECG) findings as well as genetic or familial background [21].
Previously the classification of cardiomyopathies has been based mainly on cardiac imaging or complex criteria, as for ARVC. More recently it has been observed that some phenotypes do not fit these earlier classifications. The concept of arrhythmogenic cardiomyopathy (ACM) has also been proposed, however, there are no diagnostic criteria for it so far. ACM comprises a group of cardiomyopathies with ventricular arrhythmias and right and/or left ventricular affection.
We found this DSP c.6310delA, p.(Thr2104Glnfs*12) variant interesting because it was found in a previous study in six patients who were diagnosed with DCM [13]. In this study we describe the cardiac phenotype related to this DSP variant in ten Finnish index patients and their family members.
Methods
Subjects
The study included ten index patients with the DSP variant c.6310delA, p.(Thr2104Glnfs*12), of which six participated earlier Finn-DCM study [13] and were recruited from Helsinki University Hospital and four were recruited from Tampere University Hospital. To better understand the manifestation of this DSP variant in the families, all the available family members were examined. All the participants gave written informed consent, and the study was approved by the Ethical Review Committee of The Department of Medicine, University of Helsinki (HYKS 26/99, HYKS 16/99, HYKS 17/99, HYKS 19/2000, HYKS 8/2000, Dnro 307/13/03/01/2011, TMK11§274,16.12.2015). We have the permission from Statistics Finland and Ministry of Social Affairs and Health to obtain clinical data from deceased patients for research purposes.
The probands with DCM were diagnosed with the following criteria: left ventricular end-diastolic diameter (LVEDD) > 27 mm/m2 and left ventricular systolic dysfunction (Left ventricular ejection fraction < 45%) in the absence of significant coronary artery disease or abnormal loading conditions such as hypertension or valvular disease. The relatives were also considered to be possibly affected if they had one or more of the following clinical abnormalities: conduction defects, atrial fibrillation (at age of < 50 years), sinus node dysfunction and dilatation or impaired systolic function of the left ventricle.
Family history was obtained, and pedigrees were drawn. All the available hospital records of the participants were acquired, and clinical data were collected from these records. Some of the patients were evaluated as part of this study by physical examination, 12-lead ECG, echocardiography and appropriate laboratory tests at the Heart and Lung Center in Helsinki. Cardiac magnetic resonance imaging (MRI), Holter, angiography and myocardial biopsy were performed in some cases. All participants of this study are of Finnish ethnicity.
Molecular genetic studies
The genetic testing was carried out at the Blueprint Genetics laboratory at Helsinki, Finland. Eight index patients were tested using the Blueprint Genetics Pan Cardiomyopathy Panel covering 101 genes associated with cardiomyopathies and other large NGS panels including 72 and 133 genes were each used for one patient. The presence of the DSP variant in the probands’ family members was studied by using bi-directional Sanger sequencing.
Mutation nomenclature is based on GenBank accession NM_004415.2 (DSP) with nucleotide one being the first nucleotide of the translation initiation codon ATG. The pathogenicity of this DSP variant was evaluated based on the American College of Medical Genetics and Genomics (ACMG) classification scheme [22].
Results
Genetic studies
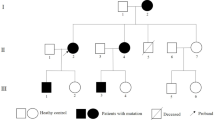
A heterozygous DSP c.6310delA, p.(Thr2104Glnfs*12) variant was observed in ten Finnish probands with cardiomyopathy. This variant causes a frameshift leading to a premature stop codon at residue 12 in a new reading frame. Thus, it is predicted to cause loss of normal protein function either through protein truncation (2114 out of 2871 aa) or nonsense-mediated mRNA decay from the other allele. It affects both RefSeq transcripts of DSP. There are 13 individuals heterozygous for this variant in the Genome Aggregation Database (gnomAD, n > 120,000 exomes and > 15,000 genomes). Of the heterozygotes, 11 are from the Finnish European cohort. Database curators have made every effort to exclude individuals with severe pediatric diseases from these cohorts. Altogether the variant was detected in 17 individuals of which 11 (65%) fulfilled DCM diagnostic criteria and 1/17 (6%) had a slightly dilated ventricle (118.8% of the estimated LVEDD using the Henry’s formula [23]). In two families the variant was observed only in the proband (Families 1 and 5, Fig. 1) and in four families (Families 6, 8, 9 and 11, Table 1) only the proband participated in the study. Pedigrees of the families (1-3 and 5-7) are shown in Fig. 1.

Pedigrees of six families affected with the DSP c.6310delA, p.(Thr2104Glnfs*12) variant. Squares represent men and circles women. Black-filled symbols represent individuals who fulfill DCM diagnostic criteria. Grey symbol represents individual who was considered affected. Arrows indicate index patients. Carriers of the DSP p.(Thr2104Glnfs*12) variant are marked in symbols with bolded outlines. Genotypes: +/− heterozygous for the DSP p.(Thr2104Glnfs*12), −/− wild type allele, * TTN p.(Val33411Thrfs*32). Year of birth, left ventricular ejection fraction and end-diastolic diameter and other clinical features listed below the symbols. LVEF left ventricular ejection fraction (%); LVEDD left ventricular end-diastolic diameter (mm). Pacemaker/Cardiac transplant: + indicates yes, − indicates no. ECG (electrocardiogram) - SR indicates sinus rhythm; (p)L/RBBB (partial) left/right bundle branch block; LAHB left anterior hemiblock. Arrhythmias - VT for ventricular tachycardia; VES for ventricular extrasystoles; AF for atrial fibrillation; NA not available. ProBNP pro b-type natriuretic peptide, ** BNP b-type natriuretic peptide
The main clinical features of the index patients and their relatives are presented in Table 1. These features include left ventricle end-diastolic diameter and ejection fraction, arrhythmias and conduction defects. Pacemaker or implantable cardioverter-defibrillator (ICD) was implanted in 3/17 (18%) of the heterozygotes and atrial fibrillation was observed in 4/17 (24%) and ventricular tachycardia or ventricular fibrillation in 5/17 (29%) of the cases. Four out of five DCM patients with severe arrhythmia episode (VT/VF) had cardiomyopathy changes prior or when first time presenting with arrhythmia episode. From one patient this information was not available. Conduction abnormalities were present in all but two of the families and atrioventricular-block (AV-block) of some degree was observed in 7/17 (41%) of the heterozygotes. Two out of 17 (12%) carriers of the DSP mutation had a heart transplantation and the other one also carried a heterozygous pathogenic TTN variant.
Nine patients underwent cardiac MRI and seven of them were carriers of the DSP variant. Of these seven, three fulfilled DCM diagnostic criteria, two had LGE as well as cardiac edema, one had only LGE. Fibrofatty replacement was not identified in any of the patients.
Two out of seven genotype positive family members had DCM at ages 57 and 60, one had dilated LV with normal EF at age 56, one had second degree AV block at age 43 and three others were considered as genotype positive yet phenotype negative at ages 40, 49 and 53. All genotype negative family members were unaffected.
Based on the variant classification scheme that follows the ACMG guidelines, this DSP c.6310delA, p.(Thr2104Glnfs*12) variant can be considered as pathogenic.
Family 1
Proband (II.5) of this family was diagnosed with DCM at the age of 51. His BNP (b-type natriuretic peptide) concentration was 2135 ng/l at its highest. He had no history of arrhythmias, but ECG showed left anterior hemiblock. On echocardiography his LVEDD was 80 mm and LVEF 10–15%. The proband later received a heart transplant. Myocardial biopsy was taken, and no amyloidosis was observed. In addition to the DSP variant, the proband also had a pathogenic variant, TTN c.100230_100234delinsGACA, p.(Val33411Thrfs*32); NM_001267550.1. The same TTN deletion, but not the DSP variant, was also found in two other members of the family, the proband’s brother (II.4) and nephew (III.7) who both were affected.
Family 2
Proband (II.2) of this family was a 67-year-old female diagnosed with DCM at the age of 46. The patient had suffered from atrial fibrillation and ECG showed right bundle branch block as well as left anterior hemiblock. Her pro-BNP (pro b-type natriuretic peptide) concentration was 650 ng/l and on echocardiography her LVEDD was 68 mm and LVEF 35%. The proband died at the age of 69 suddenly. In autopsy both cardiac ventricles were enlarged, there was no significant coronary artery disease, neither signs of myocardial infarct nor pulmonary embolism. The cause of death was considered to be cardiac. Proband’s genotype positive sister (II.4) was diagnosed with DCM at the age of 60. She has no arrhythmias, but ECG showed 1-degree AV block and left anterior hemiblock. On echocardiography her LVEDD was 55 mm and LVEF 45%. Her pro-BNP concentration was 1742 ng/l at its highest. Proband’s 49-year-old genotype positive daughter (III.2) had normal findings in cardiac evaluations. At first cardiac evaluation proband’s genotype positive 40-year-old nephew (III.6) had normal findings. One year later a cardiac MRI was performed and his LVEDV (Left Ventricular End-Diastolic Volume) was 182 ml, LVEF 54%, RVEDV (Right Ventricular End-Diastolic Volume) 173 ml and RVEF (right ventricular ejection fraction) 56%. There was considerable acute edema in the basal and mid-third anterolateral left ventricle as well as subepicardial intensive late gadolinium enhancement (LGE) within the same area.
Family 3
Proband (II.1) is an 81-year-old male diagnosed with DCM at the age of 72. He has had ventricular tachycardia as well as atrial fibrillation. ECG showed a first-degree AV block and left bundle branch block. The proband had very high levels of pro-BNP (23,478 ng/l) and on echocardiography his LVEDD was 83 mm and LVEF 18–24%. Proband’s both sons (III.1, III.2) were found to be heterozygous for the DSP variant. Proband’s eldest son (III.1) is a 56-year-old male with coronary artery disease. He has no conduction defects nor arrhythmias. On echocardiography his LVEDD was 60 mm (118.8% of the estimated LVEDD using the Henry’s formula) and LVEF 52%. Cardiac MRI demonstrated a slightly dilated left ventricle with impaired function and the wall-thickness in inferolateral area was 3–4 mm locally. Proband’s other son (III.2) is a 53-year-old male who had normal findings in cardiac evaluations.
Family 5
Proband (II.3) was diagnosed with DCM at the age of 43. She suffered from ventricular tachycardia and ventricular extrasystoles. On echocardiography her LVEDD was 60 mm and LVEF 30–35%, ECG showed a 1-degree AV block and left anterior hemiblock. Cardiac MRI demonstrated a dilated left ventricle with end-diastolic volume of 219 ml and LVEF of 41%. Her pro-BNP concentration was 1400 ng/l at its highest.
Family 6
Proband (II.1) was diagnosed with DCM at the age of 48. He had high levels on pro-BNP (9836 ng/l) and on echocardiography his LVEDD was 83 mm and LVEF 19%. He had mild mitral regurgitation and later developed a 3-degree AV block. He had ventricular tachycardia as well as atrial fibrillation and he later received a pacemaker. At the age of 59 he received a heart transplant. The proband was the only member of this family to participate in the study.
Family 7
Proband (II.2) is a 70-year-old female who developed DCM by the age of 50 years. ECG showed 2-degree AV block and left anterior hemiblock. She suffers from ventricular extrasystoles and atrial fibrillation. She has later received a CRT-D pacemaker. On echocardiography her LVEDD was 60 mm and LVEF 15–20%. Her pro-BNP concentration was 2428 ng/l at its highest. Proband’s genotype positive brother (II.5) is a 57-year-old male. ECG showed no conduction defects, but he has had ventricular extrasystoles. On echocardiography his LVEDD was 67 mm and LVEF 32%. His pro-BNP concentration was 455 ng/l. Proband’s son (III.2) died in car accident.
Family 8
Proband is a 48-year-old female, diagnosed with DCM at the age of 42. She has suffered from ventricular tachycardia and ECG showed a 1-degree AV block. She has later received a pacemaker. Echocardiography showed mild mitral regurgitation and her LVEF was 22% and LVEDD 70 mm. Cardiac MRI demonstrated a dilated left ventricle, LVEF of 20%, reduced wall-thickness and no fibrosis. Her pro-BNP was 3117 ng/l at its highest. The proband was the only member of the family to participate in the study.
Family 9
Proband is a 27-year-old female who was diagnosed with DCM at the age of 22. ECG showed no conduction defects, but she has suffered from ventricular extrasystoles. On echocardiography her LVEDD was 60 mm and LVEF 40%. At its highest her pro-BNP concentration was 400 ng/l. Cardiac MRI demonstrated an increase in signal intensity on the lateral wall of the left ventricle and there was also patchy LGE on the lateral left ventricular wall subepicardially and on the right ventricle side of the septum. Also, slightly elevated Troponin T (TnT) concentrations were observed (TnT 18–39 ng/l, normal range < 14 ng/l). Endomyocardial biopsy was taken 6 months later and it showed variation in nucleolar shape and size and myodegeneration but no sign of myocarditis. The proband was the only member of this family to participate in the study.
Family 10
Proband (III.1) was an apparently healthy 14-year-old male, who had ventricular fibrillation while watching TV at home. He was resuscitated but succumbed 2 days later at the hospital. At the hospital ECG showed no conduction defects, and on echocardiography his LVEF was 17% and LVEDD 55 mm. No known Long QT Syndrome (LQTS) causing variants were found on genetic evaluation. On autopsy, histology did not demonstrate findings compatible with acute myocarditis. There were no signs of amyloidosis or hemochromatosis. Plakoglobin staining was normal. Interstitial fibrosis of varying grades could be observed. Proband’s genotype positive father (II.1) has had ventricular extrasystoles whereas ECG showed no conduction defects. His pro-BNP concentration was 21 ng/l and on echocardiography his LVEDD was 54 mm (111% of the estimated LVEDD using the Henry’s formula) and LVEF 52%. Cardiac MRI demonstrated no clear LGE.
Family 11
Proband is a 43-year-old male with a 2-degree AV-block. Cardiac MRI demonstrated slightly dilated ventricles and atriums, LVEF was 58% and RVEF 60%. There was no edema. Linear and midmyocardial LGE compatible with fibrosis was observed. Proband was the only member of the family that could be recruited to this study and we have no knowledge of proband’s parents’ genotypes, but we do know the following considering their cause of death. Proband’s mother died suddenly at the age of 24 while pulling a sled on the street. During the 4 years that preceded her death, she had suffered from palpitations of unknown cause and she had visited the hospital several times because of chest pains. On the last of these visits, a 1-degree AV-block was observed. Proband’s father had alcoholic liver cirrhosis and he died suddenly at later age due to intoxication.
Discussion
We have identified the DSP c.6310delA, p.(Thr2104Glnfs*12) in ten Finnish index patients with DCM. This study also demonstrates relatively low penetrance of truncating DSP variant in the probands’ family members before age of 40 but phenotype became evident in about half of the family members by age 60. In addition to the previous FinnDCM study [13], this variant has previously been reported twice in literature, however, in both cases the patients were also carriers of another DSP gene mutation. In both cases the phenotype was cardio-cutaneous and in the other case, the patient suffered sudden cardiac death [24, 25].
Nine of the index patients and two relatives fulfilled the diagnostic criteria for DCM but at least one of the subjects, a young previously healthy individual, had ventricular fibrillation and SCD as the first manifestation of the disease. The age of diagnosis varied greatly as the youngest was 14 years old while the oldest participant was 72 years old when the diagnosis was made. Two of the participants presented edema on cardiac MRI. From one of these patients TnT was measured and initially elevated, then normalized in follow-up. The findings were suggestive of some kind of inflammatory process. The inflammatory process in the myocardium could explain some of the sudden arrhythmias as the disease might process periodically rather than continuously. However further studies are needed to elucidate the possible relations between DSP pathogenic variants and myocardial inflammation.
The major findings in our subjects are ventricular arrhythmias and dilatation of the left ventricle. Three of the patients died suddenly and two of them at young age. Our findings confirm the significance of DSP gene as a cause of arrhythmogenic cardiomyopathy.
Even though some of the patients do meet the DCM diagnostic criteria, it seems that the arrhythmogenic side of this phenotype is more important in practice, because the deaths were caused by arrhythmogenic events rather than ventricular dysfunction.
Desmoplakin is coded by the DSP gene on chromosome 6p24.3, and there are two isoforms produced by alternative splicing. Desmoplakin is a crucial part of forming functional desmosomes as it interacts with intermediate filaments and binds them to desmosomal plaques [15, 16]. Desmosomes are intercellular junctions that are abundant in tissues undergoing constant physical stress, such as the heart and the epidermis [15]. To adapt to their mechanical environment, cardiomyocytes may use different proteins such as integrins or strain-activated ion channels [26]. The mechanical stresses the cell experiences can be transmitted through the cytoskeleton to the nucleus by actin, intermediate filaments and microtubules [26]. The main intermediate filaments in cardiac myocytes are desmins which are connected to desmosomes. A recent study has suggested that the junction between intermediate filaments and desmosomes is specialized to endure external mechanical stress and having a stress absorbing function [27]. Desmoplakin carries a significant role in this as under most conditions it does not experience significant tension, but it is capable of sensing the exposure to external mechanical stresses and reacting to it [27].
Mutations in DSP manifest in the skin, hair and heart in humans. The participants in this study were not studied systematically for skin or hair abnormalities, but there were no mentions of these kinds of defects in the patient records. Autosomal recessively inherited mutations have previously been associated with severe disorders such as Carvajal syndrome [28,29,30], skin fragility-woolly hair syndrome and lethal acantholytic epidermolysis bullosa [31]. Autosomal dominant mutations in DSP have been associated with ARVC [32], DCM and SAM (Severe skin dermatitis, multiple allergies and metabolic wasting) syndrome [33]. There are altogether 163 truncating DSP variants reported in ClinVar (May 2019) of which 156 (95,7%) were classified as pathogenic or likely pathogenic. Most of the variants classified as non-pathogenic are located in the C-terminus.
According to gnomAD the DSP c.6310delA, p.(Thr2104Glnfs*12) frequency in Finnish population is 0.0005081. Total allele frequency is 0.00005170. The variant is enriched in Finnish population but the presence of this variant in the reference population could possibly be explained by the variant’s low penetrance before the age of 40. Because of the late expression of the phenotype, this could lead to the inclusion of variant carriers in the reference population, who have not yet developed the disease.
According to the 2008 position statement of the European Society of Cardiology, cardiomyopathies have been divided into groups based on the phenotypes and each phenotype is divided into familial and non-familial forms [1]. DCM is defined by dilatation and systolic dysfunction of the left ventricle when no abnormal loading conditions or coronary artery disease are observed [1]. Right ventricular dilatation and dysfunction may also be observed although it is not necessary for the diagnosis.
Arrhythmogenic right ventricular cardiomyopathy is a myocardial disorder defined by the dysfunction of the right ventricle and the left ventricle may also be affected. Histologically ARVC is characterized by fibrofatty replacement of the myocardium, abnormal contraction and dilatation of the right ventricle. The diagnosis is based on the latest revised Task Force diagnostic criteria, that was published in 2010 [21]. Mutations in desmosomal genes, such as PKP2 (plakophilin 2), DSP, DSG2 and DSC2, have been associated with ARVC [18]. The 2008 Task Force ARVC diagnostic criteria does not take in to account the presence of left ventricular dysfunction. The 2010 criteria also include the biventricular and LV subtypes, but there are no specific diagnostic criteria for the non-classical disease patterns [21].
Not all of our participants had their right ventricles systematically evaluated, but no abnormalities in the size or function of the right ventricle on echocardiography were reported in hospital records. Those who had cardiac MRI performed, did not show any defects in the function nor structure of the right ventricles.
This is in line with some previous studies, in which it has been noted that DSP mutations seem to be associated with left dominant or biventricular form of ARVC rather than the classical right dominant form, and DSP mutation carriers have a higher risk of SCD when compared to other mutation carriers [19, 34,35,36]. Especially the truncating mutations of DSP were associated with higher risk of developing LV dysfunction in ARVC [19].
Increasing amount of reports of clinical variants characterized by left ventricular involvement which may be parallel or greater than the RV involvement, has led to the use of a broader term of “arrhythmogenic cardiomyopathy” (ACM) [20]. Arrhythmogenic cardiomyopathy is characterized by fibrofatty replacement of the myocardium and ventricular arrhythmias as well as impairment of ventricular function. While ARVC is focused on the impairment of the right ventricle, ACM is a broader term and also includes biventricular and left-dominant subtypes [20, 37]. Arrhythmogenic left ventricular cardiomyopathy (ALVC) refers to a phenotype of left ventricular cardiomyopathy with ventricular arrhythmias, however, no diagnostic criteria have been established. As the genetic background of arrhythmogenic cardiomyopathies is gradually revealed, it will also be possible to better characterize the related phenotypic variation.
Conclusion
This s.6310delA DSP variant causes an arrhythmogenic phenotype in addition to the suboptimal function of the left ventricle although fibrofatty infiltration was not observed. Our findings concur with the proposed concept of arrhythmogenic cardiomyopathy but more research in the subject is still needed. The variant is more common in the Finnish population as compared to many other populations. It might also modify the risk of ventricular arrhythmias for example in the context of coincidental coronary artery disease. Understanding the genetic background may have some significance in the diagnosis and treatment of these diseases in the future.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due to privacy issues and GDPR legislation but are available from the corresponding author on reasonable request.
Abbreviations
- ACM:
-
Arrhythmogenic cardiomyopathy
- ACMG:
-
American College of Medical Genetics and Genomics
- ALVC:
-
Arrhythmogenic left ventricular cardiomyopathy
- ARVC:
-
Arrhythmogenic right ventricular cardiomyopathy
- AV-block:
-
Atrioventricular block
- BNP:
-
B-type natriuretic peptide
- DCM:
-
Dilated cardiomyopathy
- DES:
-
Desmin
- DSC2:
-
Desmocollin-2
- DSG2:
-
Desmoglein 2
- DSP:
-
Desmoplakin
- ECG:
-
Electrocardiogram
- FLNC:
-
Filamin C
- ICD:
-
Implantable cardioverter-defibrillator
- LGE:
-
Late gadolinium enhancement
- LMNA:
-
Lamin A/C
- LQTS:
-
Long QT Syndrome
- LV:
-
Left ventricle
- LVEDD:
-
Left ventricular end-diastolic diameter
- LVEDV:
-
Left Ventricular End-Diastolic Volume
- LVEF:
-
Left ventricular ejection fraction
- MRI:
-
Magnetic resonance imaging
- MYH7:
-
Beta-myosin heavy chain
- PKP2:
-
Plakophilin 2
- pro-BNP:
-
Pro b-type natriuretic peptide
- RBM20:
-
RNA binding motif protein 20
- RVEDV:
-
Right Ventricular End-Diastolic Volume
- RVEF:
-
Right ventricular ejection fraction
- SAM:
-
Severe skin dermatitis, multiple allergies and metabolic wasting
- SCD:
-
Sudden cardiac death
- TNNT2:
-
Troponin T2
- TnT:
-
Troponin T
- TPM1:
-
Tropomyosin 1
- TTN:
-
Titin
References
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2008;29(2):270–6 [cited 2019 May 14] Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehm342.
Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12(11):655–67 [cited 2019 may 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/20864896.
Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, et al. Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J. 1999;20(2):93–102 [cited 2019 May 14]. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1053/euhj.1998.1145.
Codd MB, Sugrue DD, Gersh BJ, Melton LJ. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation. 1989;80(3):564–572 [cited 2019 may 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/2766509.
EM MN, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123(1):19–26 [cited 2019 may 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/23281406.
Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47 [cited 2019 May 14] Available from: http://www.nature.com/articles/nrcardio.2013.105.
Cahill TJ, Ashrafian H, Watkins H. Genetic Cardiomyopathies Causing Heart Failure. Circ Res. 2013;113(6):660–75 [cited 2019 May 14] Available from: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.113.300282.
Mestroni L, MRG T. Genetics and genetic testing of dilated cardiomyopathy: a new perspective. Discov Med. 2013;15(80):43–9 [cited 2019 may 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/23375013.
Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16(8):601–8 [cited 2019 May 14] Available from: http://www.nature.com/articles/gim2013204.
Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123–35 [cited 2019 May 14]Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehu301.
Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–28 [cited 2019 may 14]Available from: http://www.ncbi.nlm.nih.gov/pubmed/22335739.
Jacoby D, WJ MK. Genetics of inherited cardiomyopathy. Eur Heart J. 2012;33(3):296–304 [cited 2019 may 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/21810862.
Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenperä P, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36(34):2327–37 [cited 2019 may 14] Available from: http://www.ncbi.nlm.nih.gov/pubmed/26084686.
McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017[cited 2019 may 14];121(7):731–748. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28912180.
Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta - Biomembr. 2008;(3):1778, 572–87 [cited 2019 May 14] Available from: https://www.sciencedirect.com/science/article/pii/S0005273607002751.
Smith EA, Fuchs E. Defining the Interactions Between Intermediate Filaments and Desmosomes. J Cell Biol. 1998 [cited 2019 may 14];141(5):1229–1241. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9606214.
Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2012[cited 2019 Jun 13];9(4):223–233. Available from: http://www.nature.com/articles/nrcardio.2011.173
Campuzano O, Alcalde M, Allegue C, Iglesias A, García-Pavía P, Partemi S, et al. Genetics of arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2013 [cited 2019 may 14];50(5):280–289. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23468208.
Castelletti S, Vischer AS, Syrris P, Crotti L, Spazzolini C, Ghidoni A, et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int J Cardiol. 2017[cited 2019 Jun 5];249:268–273. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0167527317305788
Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017 [cited 2019 Jun 3];121(7):784–802. Available from: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.117.309345
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010 [cited 2019 may 14];31(7):806–814. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20172912.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015 [cited 2019 may 14];17(5):405–424. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25741868.
Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age. Circulation. 1980;62(5):1054–61.
Vahlquist A, Virtanen M, Hellström-Pigg M, Dragomir A, Ryberg K, Wilson NJ, et al. A Scandinavian case of skin fragility, alopecia and cardiomyopathy caused by DSP mutations. Clin Exp Dermatol. 2014 [cited 2019 Jun 4];39(1):30–34. Available from: http://doi.wiley.com/10.1111/ced.12226
Mahoney MG, Sadowski S, Brennan D, Pikander P, Saukko P, Wahl J, et al. Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. J Invest Dermatol. 2010 [cited 2019 Jun 4];130(4):968–978. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19924139.
Mestroni L, Sbaizero O. Arrhythmogenic Cardiomyopathy: Mechanotransduction Going Wrong [Internet]. Vol. 137, Circulation. Lippincott Williams & Wilkins Hagerstown, MD; 2018 [cited 2019 Jul 2]. p. 1611–3. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.118.033558
Price AJ, Cost A-L, Ungewiß H, Waschke J, Dunn AR, Grashoff C. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat Commun. 2018[cited 2019 Jun 26];9(1):5284. Available from: http://www.nature.com/articles/s41467-018-07523-0
Keller D, Stepowski D, Balmer C, Simon F, Guenthard J, Bauer F, et al. De novo heterozygous desmoplakin mutations leading to Naxos-Carvajal disease. Swiss Med Wkly 2012 [cited 2019 May 14];142(3536). Available from: http://doi.emh.ch/smw.2012.13670
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Ruiz Cabezas J-C, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000 [cited 2019 May 14];9(18):2761–2766. Available from: https://academic.oup.com/hmg/article-lookup/doi/10.1093/hmg/9.18.2761
Norgett EE, Lucke TW, Bowers B, Munro CS, Leigh IM, Kelsell DP. Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantar keratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. J Invest Dermatol. 2006 [cited 2019 may 14];126(7):1651–1654. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16628197.
McGrath JA, Bolling MC, Jonkman MF. Lethal Acantholytic Epidermolysis Bullosa. Dermatol Clin. 2010 [cited 2019 May 14];28(1):131–135. Available from: https://www.sciencedirect.com/science/article/pii/S0733863509000898?via%3Dihub
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002 [cited 2019 may 14];71(5):1200–1206. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12373648.
McAleer MA, Pohler E, Smith FJD, Wilson NJ, Cole C, MacGowan S, et al. Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N-terminal plakin domain of desmoplakin. J Allergy Clin Immunol. 2015 [cited 2019 may 14];136(5):1268–1276. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26073755.
Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JDH, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015 [cited 2019 Jun 5];36(14):847–855. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehu509
Miles C, Finocchiaro G, Papadakis M, Gray B, Westaby J, Ensam B, et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation. 2019 [cited 2019 Jun 5];139(15):1786–1797. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.118.037230
López-Ayala JM, Gómez-Milanés I, Sánchez Muñoz JJ, Ruiz-Espejo F, Ortíz M, González-Carrillo J, et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype. EP Eur. 2014 [cited 2019 Jun 5];16(12):1838–1846. Available from: https://academic.oup.com/europace/article-lookup/doi/10.1093/europace/euu128
Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FC, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019 [cited 2019 Dec 26];16:e301–e372. Available from: https://doi.org/10.1016/j.hrthm.2019.05.007.
Acknowledgements
We express our gratitude to patients and their family members for their participation in this study.
Funding
This work was supported by grants from the Finnish Foundation for Cardiovascular Research (TH), Aarne Koskelo foundation (TH), Special Governmental Subsidy (EVO) grants (TH).
The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
study design: JK, TH. funding: TH, JK. recruiting participants, collecting samples or clinical data: TH, SW, KAS, SV, KH. pedigrees: SW, KH, SV, KAS. clinical cardiac evaluation: TH, SV, KAS. DNA sequencing and variant interpretation: JK, TKK, TPA, SM. analyzing results: JK, TH, KH, SW, SV, KAS, TKK, TPA, SM. drafted and wrote the manuscript: KH, JK, TH. contributed to writing of the manuscript: SW, SV, KAS, TKK, TPA, SM. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all the participants, and the study was approved by the Ethical Review Committee of The Department of Medicine, University of Helsinki (HYKS 26/99, HYKS 16/99, HYKS 17/99, HYKS 19/2000, HYKS 8/2000, Dnro 307/13/03/01/2011, TMK11§274,16.12.2015). We have the permission from Statistics Finland and Ministry of Social Affairs and Health to obtain clinical data from deceased patients for research purposes.
Consent for publication
Not Applicable.
Competing interests
Minor conflict of interest: TPA, SM, JK are co-founders and TKK, TPA, SM, JK are full-time employees of Blueprint Genetics, which offers genetic diagnostic services.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Heliö, K., Kangas-Kontio, T., Weckström, S. et al. DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy. BMC Med Genet 21, 19 (2020). https://doi.org/10.1186/s12881-020-0955-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-0955-z