
Abstract
Background
The hypothalamic G-protein-coupled-receptor melanocortin-4 receptor (MC4R) is a key player in the central circuit regulating energy expenditure and appetite. Heterozygous loss-of-function MC4R mutations are the most common known genetic cause of monogenic human obesity, with more than 200 mutations described to date, affecting 2–3% of the population in various cohorts tested. Homozygous or compound heterozygous MC4R mutations are much less frequent, and only few families have been described in which heterozygotes and homozygotes of the same mutation are found.
Methods
We performed exome sequencing in a consanguineous Bedouin family with morbid obesity to identify the genetic cause of the disease. Clinical examination and biochemical assays were done to delineate the phenotype.
Results
We report the frequency of MC4R mutations in the large inbred Bedouin Israeli population. Furthermore, we describe consanguineous inbred Bedouin kindred with multiple individuals that are either homozygous or heterozygous carries of the same novel MC4R mutation (c.124G > T, p.E42*). All family members with the homozygous mutation exhibited morbid early-onset obesity, while heterozygote individuals had either a milder overweight phenotype or no discernable phenotype compared to wild type family members. While elder individuals homozygous or heterozygous for the MC4R mutation had abnormally high triglycerides, cholesterol, glucose and HbA1C levels, most did not.
Conclusions
MC4R mutation homozygotes exhibited morbid early-onset obesity, while heterozygotes had a significantly milder overweight phenotype. Whereas obesity due to MC4R mutations is evident as of early age – most notably in homozygotes, the metabolic consequences emerge only later in life.
Similar content being viewed by others

Background
Obesity, affecting more than 30% of adults and children worldwide [1], is a complex trait affected by diet in conjunction with environmental and genetic factors [2]. The melanocortin-4 receptor (MC4R) is a key player in the leptin-regulated melanocortin circuit, essential for central energy regulation [3]. Predominantly expressed in the hypothalamus, the G-protein-coupled-receptor MC4R acts in signaling satiety, consequently decreasing food intake: upon binding of its endogenous ligand, the neuropeptide melanocyte stimulating hormone (α-MSH), MC4R activates adenylate cyclase, enhancing cAMP synthesis and levels, thereby generating a satiety signal. In line with the role of MC4R in satiety signaling and regulation of food intake, MC4R null mutant (MC4R −/−) mice develop severe obesity, while heterozygous (MC4R +/−) mice present a mildly obese intermediate phenotype [4].
Mutations in members of the leptin-regulated melanocortin circuit have been shown to result in human obesity. Most notably, human obesity has been associated with mutations in leptin (LEP) [5], leptin receptor (LEPR) [6], prohormone convertase 1 (PC1) [7], proopiomelanocortin (POMC) [8] and melanocortin-4 receptor (MC4R) [9,10,11,12,13,14,15]. In fact, heterozygous loss-of-function MC4R mutations have been shown to be the most common known genetic cause of human obesity. Initial studies by Farooqi et al. [16] estimated that about 6% of early onset morbidly obese patients harbor MC4R mutations. Consequently, other large cohort studies, mostly in Caucasian European and American populations, demonstrated lower calculated prevalence of about < 2% [17,18,19,20,21]. The prevalence of MC4R mutations in severely obese populations in other ethnicities, such as Asians, was found to be low or not relevant [16].
Homozygous or compound heterozygous carriers of MC4R mutations are rare [15, 16, 22, 23]. Previous studies suggest that the obesity in these rare cases develops earlier in life, and is more severe than for heterozygous carriers; notably, these homozygous individuals do not display any discernible additional unrelated phenotypes [16]. Only few families have been described to date in which multiple heterozygotes and homozygotes of the same mutation are found [16]. We now describe large consanguineous inbred kindred with individuals that are either homozygous or heterozygous carries of the same novel MC4R mutation, and review the literature of such families described to date.
Methods
Subjects and clinical phenotyping
Sixteen affected and unaffected individuals of consanguineous Bedouin kindred were studied (Fig. 1a). DNA samples were obtained following informed consent and approval of the Soroka Medical Center Internal Review Board (0316–14-SOR) according to Helsinki ethical guidelines. Clinical phenotyping was determined by an experienced team of pediatrics specialists and clinical geneticists for all affected individuals, their parents and siblings. Blood samples, as well as measurements of weight and height, were taken from all participants on the same day.

Pedigree of the studied kindred and the MC4R mutation. a Pedigree of a consanguineous Bedouin family presenting with a phenotype of autosomal recessive early-onset obesity (individuals with phenotypic morbid obesity are marked as affected; asterisk marks individuals whose DNA was available for analysis). b The c.124 G > T, p.(E42*) MC4R mutation: Sanger sequencing of an unaffected individual (III:7), an obligatory carrier (II:5) and a morbidly-obese affected individual (IV:1). c Schematic representation of the p.(E42*) mutation in MC4R predicted to truncate almost 90% of the protein (TM, Transmembrane domain)
Whole exome sequencing
Genomic DNA was isolated from peripheral blood using The E.Z.N.A. SQ Blood DNA Kit (Omega Bio-tek, Norcross,GA, USA). Whole exome sequencing (HiSeq2000, Illumina, San Diego, CA, USA) of two affected individuals (III:5 & IV:1, Fig. 1a) was performed using paired-end (2 × 100) protocol at a mean coverage of 100-fold (85–90% of all exonic nucleotides were covered by > 100 reads), as previously described [24]. For exome enrichment, we used NimbleGen SeqCap EZ Human Exome Library v2.0 (Roche NimbleGen, Madison, WI, USA) targeting 44.1 Mb regions. Sequencing read alignment, variant calling and annotation were performed by DNAnexus (DNAnexus Inc., Mountain View, CA, USA; dnanexus.com).
Data were analyzed using QIAGEN’s Ingenuity® Variant Analysis™ software (www.qiagen.com/ingenuity) from QIAGEN Redwood City. Using their filtering cascade, we excluded common variants which demonstrated an allele frequency higher than 0.5% in AFC (Allele Frequency Community - including ExAC and CGI), in the 1000 genomes project and in NHLBI ESP exomes (National Heart, Lung, and Blood Institute Exome Sequencing Project). In addition, we excluded variants which appeared in a homozygous state or that presented an allele frequency of higher than 2% in our in-house whole exome sequencing database of 120 Bedouin control samples. Furthermore, we kept variants which were predicted to have a deleterious effect upon protein coding sequences (e.g. Frameshift, in-frame indel, stop codon change, missense or predicted to disrupt splicing by MaxEnt Scan) and variants which were experimentally observed to be associated with a phenotype: pathogenic, possibly pathogenic or disease-associated, according to the Human Gene Mutation Database (HGMD). Following the above filtering, of the remaining variants we selected only homozygous variants that were shared between both affected individuals sequenced.
Mutation screening
Validation of the MC4R variant in all affected family members was performed by Sanger sequencing. Primers used for PCR: Forward 5’-ATCAATTCAGGGGGACACTG; Reverse 5’-AACGCTCACCAGCATATCAG. Annealing temperature used was 60 °C and the extension time was set for 30 s. Segregation analysis within the entire kindred was performed by restriction analysis using the primers mentioned above for PCR, based on an NheI restriction site generated through the MC4R c.124 G > T mutation. PCR products (207 bp amplicon) were incubated for 3 h with the enzyme NheI (New England Biolabs) which cuts selectively only the mutant allele (163 and 44 bp fragments), and subsequently loaded onto a 2% agarose gel for electrophoresis.
Results
Molecular genetic studies
In search for the molecular basis of the familial obesity, whole-exome sequencing analysis was done for two remotely related affected individuals of the kindred, namely III:5 & IV:1 (Fig. 1a). Data were filtered for normal variants as described in the materials and methods section. Following the above filtering, only a single homozygous variant was found that was shared between the two individuals: g.chr18: 58039459C < A, c.124 G > T, p.E42* in MC4R. No other variants (heterozygous or homozygous) in MC4R or in any other obesity-related gene were identified in any of the two subjects tested through whole exome sequencing. The MC4R mutation, validated by Sanger sequencing (Fig. 1b), was found to segregate within the family as expected for autosomal recessive heredity (data not shown). There is no evidence of the reported mutation being present in the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), a database of 123,136 exome sequences and 15,496 whole-genome sequences from unrelated individuals sequenced as part of various disease-specific and population genetic studies. The mutation truncates the 332-amino acid MC4R protein at amino acid 42, eliminating all its 7 transmembrane domains, hence practically all its functional domains (Fig. 1c). The mutation was not found in 120 whole exome sequences of local Bedouin controls in our in-house database.
As delineated in Table 1, of those 120 Bedouin control non-related individuals, one carried a heterozygous c.606C > A p.F202 L MC4R mutation previously associated with obesity, and two individuals carried heterozygous synonymous variants (c.594C > T, c.690C > T) previously described as possibly associated with obesity, though likely non-pathogenic [25,26,27]. Clinical obesity-related information regarding those individuals is not available. Interestingly, 4 of the 120 Bedouin controls were heterozygous for the c.307G > A p.V103I MC4R variant previously associated with protection from obesity [28] (ExAC frequency for this variant is 0.01743).
Clinical characterization
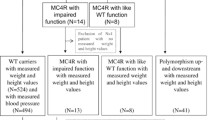
Sixteen individuals of a consanguineous Bedouin kindred were studied (ages 1–58 years, 21.7 ± 14.7 SD), of which 4 were homozygous for the MC4R mutation, 7 were heterozygous for the mutation and 5 were homozygous for the wild type sequence. As shown in Table 2, height, weight and body mass index (BMI) were measured for all, and fasting blood values of cholesterol, triglycerides, LDL, HDL, glucose and HbA1C were available for most. In cases where more than one measurement was available, the average of the measurements was calculated and is presented.
Homozygotes for the p.E42* MC4R mutation exhibited average BMI of 44.83 (44.83 ± 9.7 SD), indicative of extreme obesity (obesity class III) and significantly higher than the BMI of both heterozygous and wild type family members (p < 0.005). Individuals heterozygous for the mutation had an average BMI of 29.22 (29.22 ± 10.2 SD), within the upper limits of BMI values defined as overweight. Average BMI of family members homozygous for the wild type MC4R allele was 24.46 (24.46 ± 8.98 SD), within BMI normal values.
Fasting blood triglycerides, cholesterol, LDL, HDL, glucose and HbA1C levels were measured for most individuals studied. As seen in Table 2, only the eldest of the 4 homozygous individuals (III:5, Fig. 1a, age 30–35 years) had extremely high levels of cholesterol, triglycerides, LDL, glucose and HbA1C, while the other 3 (ages 5–10 and 25–30 years) had normal values. Similarly, of the 7 heterozygotes, high levels of triglycerides, glucose and HbA1C were found only in the eldest individual (age range 55–60 years, versus age ranges 5–10, 20–25, 25–30, 25–30, 25–30, 30–35 of heterozygotes with normal levels).
Discussion
MC4R mutations are the most common known genetic cause of obesity, affecting 2–3% of the population in various cohorts tested [16, 29]. To date, about 200 MC4R genetic variants have been identified, including at least 122 missense mutations, 2 in-frame deletion mutations, 7 nonsense mutations and dozens of frameshift mutations [3, 30], altogether affecting more than 30% of the receptor coding sequence. While it has been suggested that obesity due to MC4R mutations can be caused by either haplo-insufficiency or dominant negative activity exerted by the mutant receptor, co-transfection studies show that the extreme majority of mutations analyzed do not have dominant negative activity [3, 15, 31, 32]. Therefore, it is suggested that haplo-insufficiency is the main route through which these mutations exert their effect. While loss-of-function mutations in MC4R cause familial forms of obesity, two rare gain-of-function MC4R polymorphisms have been identified that are associated with protection against obesity [19]. In fact, we show that in our cohort of 120 control Bedouin whole exome sequences, 4 individuals are heterozygous for one of these variants, namely c.307G > A, p.V103I. Meta analysis of previously published data showed that this gain-of function mutation has a modest negative association with obesity [19]. It is of interest that the prevalence of the p.V103I variant in the Israeli Bedouin community seems to be higher than worldwide (ExAC frequency 0.01743). Notably, another gain-of-function c.751A > C p.I251L MC4R variant, that is more clearly negatively associated with obesity worldwide [19], was not found in our Bedouin cohort. Obviously, larger cohorts within this large inbred Bedouin community [33] should be tested to validate statistical significance of these observations.
In families with MC4R-associated obesity, obesity tends to have an autosomal dominant mode of transmission, but the penetrance of the disease can be incomplete and the clinical expression variable (moderate to severe obesity), underscoring the role of the environment and other possible modulating genetic factors [34, 35]. As heterozygous MC4R mutation carriers are obese, yet present with partial penetrance of the mutations, O’Rahilly and colleagues concluded that the mode of inheritance in MC4R deficiency is codominance with modulation of expressivity and penetrance of the phenotype [36]. It has been suggested that the varying onset and severity of obesity in heterozygous MC4R mutation carriers are related to the severity of the functional effects of the mutations. In fact, with many human MC4R mutations identified, several research groups (Tao et al. [3], MacKenzie [29], Vaisse et al. [14], Farooqi et al. [15]) classified the MC4R mutations based on possible functional consequences: mutations that cause intracellular-retaining of the receptor, defective expression, defective binding, defects in both basal and ligand-induced signaling, etc. However, as only a minority of the variants underwent in-depth functional analysis, validity of such classifications in the context of clinical phenotypic association awaits further studies.
Homozygous or compound heterozygous carriers of MC4R mutations are rare [15, 22, 23]. Only few families have been described to date in which multiple heterozygotes and homozygotes of the same mutation are found. We now identified a novel MC4R truncation mutation, putatively abolishing all 7 transmembrane domains of the molecule (Fig. 1c). Previous studies reported phenotypic variation in consequences of heterozygous MC4R deletion mutations [19, 20]. As the cohort we studied is small, while the average BMI in heterozygous individuals was higher than in wild type family members (29.22 versus 24.24.46, respectively), this difference was not statistically significant, neither in adults nor in children.
Unique to our study, we delineated the mutation-related phenotype in large consanguineous kindred with 4 homozygous and 7 heterozygous individuals, as well as 5 wild type family members. This unique kindred, of few identified thus far, allows insights as to phenotypic effects in heterozygotes versus homozygotes of the same mutation. As evident in Table 2, although the cohort is too small to establish statistical significance, the data clearly point to early-onset obesity in individuals homozygous for the MC4R mutation: of the children (ages 1–7 years) within the kindred, the two homozygotes were morbidly obese (BMI 37 and 37; Z scores 3.12, 3.08) as compared to the two wild type individuals (BMI 18 and 17) and the heterozygous individual (BMI 17) that were within normal BMI values (85th BMI-per age percentile). In fact, in spite of the fact that a single kindred is described, the clear morbid obesity (average BMI 44.83) in homozygous individuals is statistically significant compared to the overweight (average BMI 29.22) in heterozygous individuals and the higher end of normal weight (average BMI 24.46) seen in wild type family members. This is in line with previous reports, showing a dramatically more consistent and severe obesity phenotype in homozygotes for MC4R mutations than in heterozygotes [16]. Furthermore, the obesity in the homozygotes is of early onset, with BMI Z scores ~ 3 in individuals ages 5–10 years. While previous studies of MC4R heterozygotes have shown age-dependent differences in expressivity and a stronger effect in females [16, 18,19,20,21, 34], the cohort in the present study is too small to reach conclusions in this regard. Interestingly, although all affected individuals share the same mutation and reside in the same environment (practically the same household), there is variability in phenotypic expression, in the heterozygous individuals in particular, suggesting possible effects of modifier genes. In fact, future studies of such kindreds might be conducive to elucidation of such modifiers.
Previous reports of homozygotes vs heterozygotes of the same MC4R mutations did not systematically describe related blood biochemistry values. Fasting blood triglycerides, cholesterol, LDL, HDL, glucose and HbA1C levels were measured for most individuals in our studied kindred. As seen in Table 2, only the eldest of the 4 homozygous individuals (III:5, Fig. 1a, age 30–35 years) had extremely high levels of cholesterol, triglycerides, LDL, glucose and HbA1C, while the other 3 (ages 5–10 and 25–30 years) had normal values. Similarly, of the 7 heterozygotes, high levels of triglycerides, glucose and HbA1C were found only in the eldest individual (age range 55–60 years vs age ranges 5–10, 20–25, 25–30, 25–30, 25–30, 30–35).
Conclusions
Through studies of a large inbred kindred we demonstrate a novel MC4R mutation, practically eliminating all functional domains of the encoded protein. We show that the phenotype in homozygotes for the mutation is significantly more severe than that of heterozygous carriers of the same mutation. While obesity, as delineated through BMI measurements, is evident in homozygotes (yet not necessarily in heterozygotes) at early ages, the metabolic consequences of MC4R-related obesity appear in both heterozygotes and homozygotes only at later ages.
Abbreviations
- BMI:
-
Body mass index
- LEP:
-
Leptin
- LEPR:
-
Leptin receptor
- MC4R:
-
Melanocortin-4 receptor
- PC1:
-
Prohormone convertase 1
- PCR:
-
Polymerase chain reaction
- POMC:
-
Proopiomelanocortin
References
Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–89.
Braunschweig, G. & Fantuzzi, C. Adipose Tissue and Adipokines in Health and Disease. (Springer, 2014). doi:https://doi.org/10.1007/978-1-62703-770-9
Tao Y-X, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544–51.
Dubern B. MC4R and MC3R Mutations. In Frelut ML, editor. The ECOG’s eBook on Child and Adolescent Obesity. 2015. Retrieved from ebook.ecog-obesity.eu
Montague CT, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8.
Clement K, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401.
Jackson RS, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6.
Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A. Severe Early-Onset Obesity, Adrenal Insufficiency and Red Hair Pigmentation Caused by POMC Mutations in Humans. Nat. Genet. 1999;19:155–7.
Yeo GSH, et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–2.
Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–4.
Gu W, et al. Identification and functional analysis of novel human melanocortin-4 receptor variants. Diabetes. 1999;48:635–9.
Hinney A, et al. Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J Clin Endocrinol Metab. 1999;84:1483–6.
Sina M, et al. Phenotypes in three pedigrees with autosomal dominant obesity caused by haploinsufficiency mutations in the melanocortin-4 receptor gene. Am J Hum Genet. 1999;65:1501–7.
Vaisse C, et al. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–62.
Farooqi IS, et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271–9.
Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95.
Hinney A, et al. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab. 2003;88:4258–67.
Valli-Jaakola K, et al. Identification and characterization of melanocortin-4 receptor gene mutations in morbidly obese finnish children and adults. J Clin Endocrinol Metab. 2004;89:940–5.
Stutzmann F, et al. Prevalence of melanocortin-4 receptor deficiency in europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes. 2008;57:2511–8.
Roth CL, et al. A novel melanocortin-4 receptor gene mutation in a female patient with severe childhood obesity. Endocrine. 2009;36:52–9.
Calton MA, et al. Association of functionally significant Melanocortin-4 but not Melanocortin-3 receptor mutations with severe adult obesity in a large north American case-control study. Hum Mol Genet. 2009;18:1140–7.
Lubrano-Berthelier C, Le Stunff C, Bougnères P, Vaisse C. A homozygous null mutation delineates the role of the Melanocortin-4 receptor in humans. J Clin Endocrinol Metab. 2004;89:2028–32.
Dubern B, et al. Homozygous null mutation of the Melanocortin-4 receptor and severe early-onset obesity. J Pediatr. 2007;150:613–8.
Perez Y, et al. UNC80 mutation causes a syndrome of hypotonia, severe intellectual disability, dyskinesia and dysmorphism, similar to that caused by mutations in its interacting cation channel NALCN. J. Med. Genet. jmedgenet-2015-103352. 2015; https://doi.org/10.1136/jmedgenet-2015-103352.
Rettenbacher E, et al. A novel non-synonymous mutation in the melanocortin-4 receptor gene (MC4R) in a 2-year-old Austrian girl with extreme obesity. Exp Clin Endocrinol Diabetes. 2007;115:7–12.
Logan M, et al. Allelic variants of the Melanocortin 4 receptor ( MC4R ) gene in a south African study group. Mol Genet Genomic Med. 2016;4:68–76.
van den Berg L, et al. Melanocortin-4 receptor gene mutations in a Dutch cohort of obese children. Obesity. 2011;19:604–11.
Young EH, et al. The V103I polymorphism of the MC4R gene and obesity: population based studies and meta-analysis of 29 563 individuals. Int J Obes (Lond). 2007;31:1437–41.
MacKenzie RG. Obesity-associated mutations in the human melanocortin-4 receptor gene. Peptides. 2006;27:395–403.
Loos RJF, et al. Melanocortin-4 receptor gene and physical activity in the Québec family study. Int J Obes. 2005;29:420–8.
Yeo GSH, et al. Mutations in the human melanocortin-4 receptor gene associated with severe familial obesity disrupts receptor function through multiple molecular mechanisms. Hum Mol Genet. 2003;12:561–74.
Ho G, MacKenzie RG. Functional characterization of mutations in melanocortin-4 receptor associated with human obesity. J Biol Chem. 1999;274:35816–22.
Markus B, Alshafee I, Birk OS. Deciphering the fine-structure of tribal admixture in the Bedouin population using genomic data. Heredity (Edinb). 2013;112:182–9.
Lubrano-Berthelier C, et al. Melanocortin 4 receptor mutations in a large cohort of severely obese adults: prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating. J Clin Endocrinol Metab. 2006;91:1811–8.
Hinney A, et al. Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population-based sample and obese adults from Germany. J Clin Endocrinol Metab. 2006;91:1761–9.
O’Rahilly S, Sadaf Farooqi I, Yeo GSH, Challis BG. Minireview: human obesity - lessons from monogenic disorders. Endocrinology. 2003;144:3757–64.
Acknowledgements
We thank the kindred participating in the study.
Funding
Funding for this research was provided by the Legacy Heritage Bio-Medical Program of the Israel Science Foundation (grant no. 1520/09) awarded to Prof. Ruth Birk and Prof. Ohad Birk.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the Sequence Read Archive (SRA) repository, https://www.ncbi.nlm.nih.gov/bioproject/478831.
Author information
Authors and Affiliations
Contributions
MD did the experiments, interpreted data and contributed to writing the manuscript. OSB interpreted data and contributed to writing the manuscript. RB initiated the study, interpreted data and wrote the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Studies were done following informed consent and approval of the Soroka Medical Center Internal Review Board (0316–14-SOR) according to Helsinki ethical guidelines. Participation of minors in the study was done with their assent and with written consent by their parents / legal guardians.
Consent for publication
not applicable.
Competing interests
Prof. Ohad Birk is an associate editor of BMC Medical Genetics. The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Drabkin, M., Birk, O.S. & Birk, R. Heterozygous versus homozygous phenotype caused by the same MC4R mutation: novel mutation affecting a large consanguineous kindred. BMC Med Genet 19, 135 (2018). https://doi.org/10.1186/s12881-018-0654-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0654-1