
Abstract
Background
The contribution of moderate coagulation factor XII (FXII) deficiency to development of thromboembolism is still undetermined. We have tried to show the relevance of FXII deficiency to incidences of venous thrombosis by exploring the prevalence of F12 gene mutations in Chinese patients with thrombotic disorders.
Methods
One hundred and six patients with venous thromboembolism (VTE) and 220 healthy controls were enrolled in study. The coding region and flanking sequences of F12 gene were amplified and sequenced to identify genetic variances. Patients with F12 mutations were also screened for other thrombotic risk factors.
Results
Heterozygous F12 gene mutations were identified in 6 individuals with VTE and 10 healthy controls. Q336X and R66W were found in two healthy individuals; D291E was identified in a patient with DVT; and A343P was a recurrent mutation with a prevalence of 4.7% (5/106) in patient group and 3.6%(8/220) in healthy control. The prevalence of heterozygous mutations between the two groups had no significant difference. The association of A343P mutations with VTE was weak with an OR of 1.31 (95% CI 0.42-4.11). No other thrombophilia risk factors screened were positive in patients harboring heterozygous F12 mutations.
Conclusions
There were conflicting theories about the relationship between FXII deficiency and thrombosis formation. Heterozygous F12 mutation decreases the plasma FXII activity approximately by half and cause moderate FXII deficiency. Although multiple mutations were identified in both groups, the link between F12 heterozygous mutation and development of thrombotic disorders is weak and further studies are warranted to clarify their relationship.
Similar content being viewed by others


Background
Factor XII (FXII) is a part of contact activation pathway of blood coagulation and lack of it will lead to prolongation of blood clotting time initiated with artificial negatively charged surfaces [1]; however, patients with congenital FXII deficiency did not experience increased bleeding diathesis and some even suffered from thrombotic disorders [2]. Thus the role of FXII plays in blood coagulation has been an intriguing question since its discovery in 1950s with conflicting results from multiple clinical and experimental studies [3, 4]. The huge variation of plasma FXII level among normal populations complicates efforts to find out the relationship between FXII activity and pathogenesis of thrombosis. Since factor XII coding gene (F12) mutations will inevitably lead to decrease of FXII activity and are definite causes for FXII deficiency [5], we tried to explore the FXII deficiency in thrombosis disorders by screening F12 mutations instead of measuring FXII activity.
Methods
Patients
Patients seeking bleeding and thrombotic disorders consultation in Ruijin Hospital, Shanghai, China, from 2012 through 2015 were screened. Totally 106 patients, including 68 males and 38 females (average age of 39.5 years, range 13~ 79), with venous thromboembolism (VTE) occurred in an early age (≤45 Years old) or recurrent VTE (≥2 times) without obvious provoking conditions, such as immobilization, using oral contraceptive, pregnancy, etc., were enrolled for studies. Blood sample was acquired via venipuncture and treated with 0.03% sodium citrate and leukocyte and plasma were separated by centrifugation at 2000 g for 15 min. Plasma samples were frozen at − 80 °C till further analysis and genomic DNA was extracted from isolated leukocytes using QIAamp DNA mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Blood samples from 220 adults, constituted with 133 males and 87 females with an average age of 36.6(range 18~ 70), without history of thrombotic or bleeding complications were taken as normal control in current study. The study was approved by ethic committee of Ruijin Hospital affiliated Shanghai Jiao tong University and informed consents were obtained from all subjects enrolled.
Genetic analysis of the F12 gene
All exons of F12 gene and noncoding sequences on either side of the coding region were amplified by PCR and sequenced directly. The genetic variation was identified by aligning sequencing results against F12 gene reference sequence (NG_007568.1) in NCBI database. DNA samples from 220 healthy controls were also screened for F12 gene mutation following same protocol mentioned above. The prevalences of F12 mutations in patients with thrombosis and normal controls were compared.
The established genetic risk factors for thrombosis, including Factor V leiden mutation and prothrombin G20210A mutation were also tested among patients carrying F12 gene mutations using PCR-RFLP methods as previously described [6].
Coagulation assays
Citrate anti-coagulated plasma was subjected to a panel of assays to assess functional status of blood coagulation systems of patients with F12 gene mutations. The traditional coagulation tests, such as activated partial thromboplastin time (aPTT) and prothrombin time (PT), were performed as first-line screening tests.
The plasma FXII activities (FXII:C) of patients with F12 gene mutations were measured by a one-stage clotting assay using FXII deficient plasma on an ACL-TOP automatic coagulometer (Instrumentation Laboratory, Bedford, MA). Other common thrombotic risk factors,such as deficiencies of antithrombin (AT), protein C (PC) and Free protein S (FPS), presence of antiphospholipid antibodies and increased homeocysteine (Hcy) level, were also screened. Briefly, activities of AT and PC were detected using chromogenic substrate methods (Instrumentation Laboratory, Bedford, MA). Free PS antigen level was measured with the ZYMUTEST Free protein S kit (Hyphen Biomed, Neuville-Sur-Oise, France). Lupus anticoagulant was analyzed by a diluted viper venom time (DVVT) assay (Instrumentation Laboratory, Bedford, MA). The anti-cardiolipin antibody (ACA) and anti-β2 glycoprotein I (anti-β2GPI) levels were detected by ELISA (Euroimmun, Lübeck, Germany). Hcy levels were measured using the AxSYM homocysteine kit (Abbott, Lake County, IL, USA).
F12 C46T variant detection
Common F12 C46T variant was detected using polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) method according to conditions described by Zito et al. [7]. The prevalences of genotype C/C, C/T and T/T were determined in both patients and healthy control group.
Statistical analysis
The difference between the prevalences of F12 heterozygous mutations in VTE and healthy control groups were analyzed by Chi-square test. Logistic regression analysis was adopted to estimate associations between recurrent heterozygous F12 mutation and VTE risk. Odds ratios (ORs) and 95% confidence intervals (95% CI) were calculated from the logistic model. The statistical analyses were performed with SPSS 19.0 software (SPSS, Chicago, IL, USA).
Results
Identification of heterozygous FXII mutations in thrombophilia patients
Multiple heterozygous F12 gene mutations were identified in both groups. Two variants (A343P and D291E) were identified in 6 individuals of the VTE group (6/106, 5.7%);three variants (A343P, Q336X and R66W) were found in 10 individuals of the healthy control group (10/220, 4.5%). The prevalence of heterozygous individuals had no significant difference between the two groups (p = 0.6625). Mutations D291E, Q336X and R66W were each only identified once in a single subject; while the A343P mutation was recurrently found in 5 unrelated patients (5/101, 4.7%) and 8 healthy controls (8/220, 3.6%). The association of A343P mutation and VTE risk was low with an OR of 1.31 (95% CI 0.42-4.11).
Coagulation assays
Patients harboring F12 mutations included 4 males and 2 females, aging from 23 through 58 years old, and none of them had concurrence of other established risk factor for VTE. The F12 gene mutations only led to moderately decreased plasma FXII:C, ranging from 33.2 to 73% (Table 1).
Prevalence of F12 C46T variant in patients with VTE and healthy control
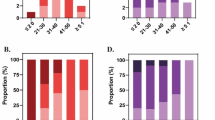
There is no significant difference of the prevalences of F12 C46T variant genotypes between patients with thrombotic events and normal healthy control (p = 0.9633). Patients and healthy control group shared similar prevalences of three genotypes C/C (56.4%, 57.2%), C/T (41.5%, 40.4%)and TT (2.1%, 2.4%), with C/C and C/T dominating in both groups. (Fig. 1).

Prevalences of F12 C46T variant in patients with venous thromboembolism and healthy control. The three genotypes C/C, C/T and TT have similar prevalences between patients with thrombotic complications and normal healthy control (C/C 56.4% vs 57.2%; C/T 41.5% vs 40.4%; T/T 2.1% vs 2.4%). The genotypes C/C and C/T are dominant in both groups
Discussion
FXII is synthesized and secreted from liver in its zymogen form and is converted by autoactivation into its own enzymatically active form, FXIIa, on negatively charged surfaces, such as dextran sulfate, kaoline, etc [8]. FXIIa catalyzes prekallikrein (PK) and factor XI (FXI) to initiate the contact pathway of blood coagulation cascade; in the meantime, it helps breaking down of fibrin clot by converting plasminogen to plasmin and starting fibrinolysis [9]. FXIIa may also up-regulate directly or indirectly various mediators involved in the inflammation response [10].
FXII is encoded by F12 gene on chromosome 5 and congenital FXII deficiency is a rare autosomal recessive disease with an estimated prevalence around 0.3% [11]. FXII deficiency will delay fibrin clot formation under artificial blood coagulation tests, however, it is mostly asymptomatic and almost no patients suffered from excessive bleeding. Counterintuitively, some even experience thrombosis.
Thrombosis model of mice lacking coagulation FXII showed that although FXII was not essential to physiological hemostasis, it might contribute to aggravation of thrombosis [12]. There were conflicting reports on FXII deficiency and thrombosis in epidemiological studies. Halbmayer WM et al. found that among 103 patients with recurrent venous thrombosis, 8% had reduced FXII level, suggesting that lack of FXII could lead to thromboembolism [13]. Another study performed in Caucasian also revealed higher prevalence of FXII deficiency in patients with DVT/PE (8.0%) than normal population (2.3%), supporting the notion that FXII deficiency might constitute a risk factor for thrombosis [14]. However, Koster, Ted et al. showed the prevalence of factor XII deficiency in patients suffering DVT/PE was not different from that of normal healthy control (6% vs 5%) [3].
The general population has a large range of plasma FXII level, with a normal range from around 80 to 120% [14, 15]. FXII level could be affected by multiple factors. There is racial difference [16], and oriental people might have lower FXII level than Caucasians; some SNPs within F12 gene were also suggested to influence expression of FXII. Previous studies used different criteria for diagnosis of moderate FXII deficiency, with cutoff FXII level ranging 20~ 57% [3, 11, 14], which might confound the relationship between moderate plasma FXII deficiency and thrombosis.
Genetic polymorphism, such as F12 C46T variant, was suggested to influence plasma FXII level [17], and it might be associated with decreased FXII activity and correlated with increased risk of thrombosis [18]. However, our result does not support this finding.
F12 gene mutation impairs FXII expression and is the definitive cause for FXII deficiency. The homozygous F12 mutation leads to complete deficiency of FXII. The heterozygous F12 gene mutation reduces FXII level by half and the FXII:C level falls in a wide range between 20 and 60%, which overlaps with normal reference FXII level (50~ 150%). It could be difficult to distinguish between individuals bearing F12 heterozygous mutations with healthy subjects who just happen to have a FXII level at lower end of normal reference range.
In current study, we screened F12 gene of patients with unprovoked VTE and tried to evaluate the risk of thrombosis in those who had their FXII activity compromised because of F12 mutation. Comparing with plasma FXII activity, F12 gene variance identified is a more accurate indicator for moderate FXII deficiency. Our results showed similar prevalence of heterozygous F12 mutations, including a hot spot mutation Ala343Pro, in thrombophilia patients and normal population. The FXII mutation Ala343Pro is characterized by inconsistent decrease of FXII clotting activity and antigen level in plasma. First identified in Japanese by Iijima, K.et [19], Ala343Pro has different prevalences among populations, and is overrepresented in East Asian races according to 1000 genome project. Animal studies suggested that FXII did not contribute to the physiological hemostasis, instead, it played a critical role in the pathological thrombosis formation. Significantly decreased FXII level (< 25%) could limit development of thrombosis in mice [20], however moderate FXII deficiency caused by heterologous F12 mutation might not be able to protect thrombosis formation.
Anticoagulation therapy targeting FXII is both effective and safe, lessening risk of bleeding afflicted by most blood thinner used today, but sufficient suppression of FXII may be essential to the success of thrombosis prevention.
Conclusions
Current study for the fist time reveals the prevalence of F12 heterologous mutations in Chinese population and shows that there is no or only weak relationship between incidents of thrombosis and moderate FXII deficiency caused by heterozygous F12 mutations.
Abbreviations
- ACA:
-
Anti-cardiolipin antibody
- anti-β2GPI:
-
Anti-β2 glycoprotein I
- APS-Ab:
-
Anti-phospholipid syndrome-associated antibodies
- aPTT:
-
Activated partial thromboplastin time
- AT:A:
-
Antithrombin activity
- CVST:
-
Cerebral venous sinus thrombosis
- DVT:
-
Deep vein thrombosis
- DVVT:
-
Diluted viper venom time;
- FPS:Ag:
-
Free protein S antigen
- FXI:
-
Coagulation factor XI
- FXII:
-
Coagulation factor XII
- FXII:C:
-
Factor XII activity
- Hcy:
-
Homocysteine
- ORs:
-
Odds ratios
- PC:A:
-
Protein C activity
- PCR-RFLP:
-
Polymerase chain reaction-restriction fragment length polymorphism
- PE:
-
Pulmonary embolism
- PK:
-
Prekallikrein
- PT:
-
Prothrombin time
- VTE:
-
Venous thromboembolism
References
Ramot B, Singer K, Heller P, Zimmerman HJ. Hageman factor (HF) deficiency. Blood. 1956;11(8):745–52.
Lämmle BWW, Huber I, Krauskopf M, Zürcher C, Pflugshaupt R, Furlan M. Thromboembolism and bleeding tendency in congenital factor XII deficiency--a study on 74 subjects from 14 Swiss families. Thromb Haemost. 1991;65(2):117–21.
Koster T, Rosendaal FR, Briet E, Vandenbroucke JP. John Hageman’s factor and deep-vein thrombosis: Leiden thrombophilia study. Br J Haematol. 1994;87(2):422–4.
Girolami A, Randi ML, Gavasso S, Lombardi AM, Spiezia F. The occasional venous thromboses seen in patients with severe (homozygous) FXII deficiency are probably due to associated risk factors: a study of prevalence in 21 patients and review of the literature. J Thromb Thrombolysis. 2004;17(2):139–43.
Schloesser M, et al. Mutations in the human factor XII gene. Blood. 1997;90(10):3967–77.
Hadhri S, et al. Factor V Leiden, prothrombin 20210G>a, MTHFR 677C>T and 1298A>C, and homocysteinemia in Tunisian blood donors. J Clin Lab Anal. 2012;26(3):167–73.
Zito F, et al. Epidemiological and genetic associations of activated factor XII concentration with factor VII activity, fibrinopeptide a concentration, and risk of coronary heart disease in men. Circulation. 2000;102(17):2058–62.
Griffin JH. Role of surface in surface-dependent activation of Hageman factor (blood coagulation factor XII). Proc Natl Acad Sci. 1978;75(4):1998–2002.
Konings J, et al. The role of activated coagulation factor XII in overall clot stability and fibrinolysis. Thromb Res. 2015;136(2):474–80.
Jansen P, et al. Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin-6 and neutrophil elastase. Blood. 1996;87(6):2337–44.
Halbmayer WMHA, Schön R, Mannhalter C, Strohmer E, Baumgarten K, Fischer M. The prevalence of moderate and severe FXII (Hageman factor) deficiency among the normal population: evaluation of the incidence of FXII deficiency among 300 healthy blood donors. Thromb Haemost. 1994;71(1):68–72.
Renné T, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–81.
Halbmayer WMMC, Feichtinger C, Rubi K, Fischer M. The prevalence of factor XII deficiency in 103 orally anticoagulated outpatients suffering from recurrent venous and/or arterial thromboembolism. Thromb Haemost. 1992;68(3):285–90.
Gallimore MJ, Harris SL, Jones DW, Winter M. Plasma levels of factor XII, prekallikrein and high molecular weight kininogen in normal blood donors and patients having suffered venous thrombosis. Thromb Res. 2004;114(2):91–6.
Calafell F, et al. Sequence variation and genetic evolution at the human F12 locus: mapping quantitative trait nucleotides that influence FXII plasma levels. Hum Mol Genet. 2010;19(3):517–25.
Gordon EM, Donaldson VH, Saito H, Su E, Ratnoff OD. Reduced titers of Hageman factor (factor XII) in Orientals. Ann Intern Med. 1981;95(6):697–700.
Soria JM, et al. A quantitative-trait locus in the human factor XII gene influences both plasma factor XII levels and susceptibility to thrombotic disease. Am J Hum Genet. 2002;70(3):567–74.
Endler G, et al. A common C→T polymorphism at nt 46 in the promoter region of coagulation factor XII is associated with decreased factor XII activity. Thromb Res. 2001;101(4):255–60.
Iijima K, Arakawa Y, Sugahara Y, Matsushita M, Moriguchi Y, Shimohiro H, Nakagawa M. Factor XII Osaka: abnormal factor XII with partially defective prekallikrein cleavage activity. Thromb Haemost. 2011;105(3):473–8.
Heestermans M, et al. Role of platelets, neutrophils, and factor XII in spontaneous venous thrombosis in mice. Blood. 2016;127(21):2630–7.
Acknowledgements
We are grateful to all the subjects enrolled in the study.
Funding
This work is supported by National Natural Science Foundation of China NSFC 81670130 (W.W.).
Availability of data and materials
All data generated or analyzed during this study are either included in this published article or are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the manuscript. WW and JD designed experiments, analyzed data, and wrote the paper; X.W and JD performed the clotting function assays and F12 gene analysis of patients; XW and QD established the diagnosis of the patients, over sighted experiments and wrote the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by ethic committee of Ruijin Hospital affiliated Shanghai Jiao tong University and informed consent were obtained from all subjects enrolled or legal guardians of individuals under age 16.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, X., Ding, Q., Wang, X. et al. The prevalence of heterozygous F12 mutations in Chinese population and its relevance to incidents of thrombosis. BMC Med Genet 19, 50 (2018). https://doi.org/10.1186/s12881-018-0557-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0557-1