
Abstract
Background
Non-ketotic hyperglycinemia (NKH) is a rare, devastating autosomal recessive disorder of glycine metabolism with a very poor prognosis. Currently, few studies have reported genetic profiling of Chinese NKH patients. This study aimed to identify the genetic mutations in a Chinese family with NKH.
Methods
A Chinese family of Han ethnicity, with three siblings with NKH was studied. Sanger sequencing and multiplex ligation-dependent probe amplification combined with SYBR green real-time quantitative PCR was used to identify potential mutations in the GLDC, AMT and GCSH genes. The potential pathogenicity of the identified missense mutation was analyzed using SIFT, PolyPhen-2, PROVEAN and MutationTaster software.
Results
All patients exhibited severe and progressive clinical symptoms, including lethargy, hypotonia and seizures, and had greatly elevated glycine levels in their plasma and CSF. Molecular genetic analysis identified compound heterozygous variants in the GLDC gene in these three siblings, including a novel missense variant c.2680A > G (p.Thr894Ala) in exon 23 and a heterozygous deletion of exon 3, which were inherited respectively from their parents. In silico analysis, using several different types of bioinformatic software, predicted that the novel variant c.2680A > G in the GLDC gene was pathogenic. Moreover, the deletion of exon 3 was identified for the first time in a Chinese population.
Conclusions
A novel missense variant and a previously reported deletion in GLDC gene were identified. The two variants of GLDC gene identified probably underlie the pathogenesis of non-ketotic hyperglycinemia in this family, and also enrich the mutational spectrum of GLDC gene.
Similar content being viewed by others
Background
Non-ketotic hyperglycinemia (NKH; OMIM 605899), also known as glycine encephalopathy, is an autosomal recessive metabolic disorder caused by a deficiency in the glycine cleavage system (GCS), resulting in a massive accumulation of glycine in body fluids [1]. Most patients present with lethargy and hypotonia in the first week of life, and often progress to apnea requiring ventilation [2]. Some patients die during the neonatal period. The majority of survivors exhibit severe mental retardation and intractable seizures typical of severe NKH [3]. One-sixth of NKH patients have an attenuated form of the disease, half of whom present in early-to-mid infancy with seizures, hypotonia, and developmental delay and/or cognitive impairments, behavioral problems, and impaired work or school performance [4]. Diagnosis is based on the detection of elevated glycine concentrations in cerebrospinal fluid (CSF) together with an increased CSF/plasma glycine ratio.
The GCS consists of the enzymes glycine decarboxylase (P-protein), amino-methyltransferase (T-protein), hydrogen carrier protein (H-protein), and dihydrolipoamide dehydrogenase (L-protein) [5]. The P, T, and H proteins are encoded by GLDC (OMIM 238300), AMT (OMIM 238310), and GCSH (OMIM 238330) genes, respectively. Approximately 70~75% of affected individuals carry disease-causing mutations in GLDC gene, whereas 20% and <1% NKH patients have mutations in AMT and GCSH genes, respectively [6]. In addition, approximately 5% of patients with enzyme-proven glycine encephalopathy do not have a pathogenic variant in GLDC, AMT or GCSH genes; these cases therefore represent a variant form of NKH [7]. Although forming only a relatively small percentage of NKH cases, in a populous country, such as China, it is possible that NKH is much more prevalent in China than it has been realized. Only a few studies have, however, examined the genetic profile of NKH patients in China, with the result that only two Chinese NKH patients have been diagnosed using molecular genetic analyses [8, 9]. We here report the clinical and genetic features of a Chinese family including three siblings with NKH.
Methods
Subjects
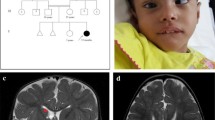
This study investigated a two-generation Chinese family containing six members of Han ethnicity (Fig. 1a). Moreover, a total of 100 healthy newborns with normal results of neonatal disease screening by tandem mass spectrometry from our center were recruited as controls, these control groups with normal phenotype and have no family history of inherited metabolic diseases including non-ketotic hyperglycinemia. The study was approved by the ethics committee of The Women’s and Children’s Hospital of Quanzhou. All the parents of the patients and 100 control subjects signed written informed consent to participate in the study, and using their own and children’s genetic data, clinical details and/or any accompanying images for scientific research and publication.

a Pedigree of the family. The filled black symbols represent the affected members and the arrow denotes the proband. b Diffusion Weight Imaging (DWI) of MRI showed extensive white matter diffusion restriction extending to the subcortical white matter in the proband’s younger sister. White matter shows a hyper-intense signal by DWI. c-e Sequence analysis of GLDC gene separately identified the heterozygous c.2680A > G variant in the proband (c) and his mother (d), but not in his father (e). f Amino acid alignment of the P-protein from several organisms. The position of Thr894 residue (highlighted by a red box) was highly conserved among different species
DNA extraction and Sanger sequencing
Genomic DNA was extracted from peripheral whole blood or dried blood spots obtained from the proband and his family members, as well as control subjects. The coding region and flanking intron sequences of GLDC (NM_000170.2), AMT (NM_000481.3) and GCSH (NM_004483.4) genes were amplified using standard PCR (polymerase chain reaction) conditions and bi-directional DNA sequencing. All the primers used are listed in Additional file 1: Table S1. The PCR cycle consisted of an initial denaturation step of 2 min at 95 °C followed by 36 cycles of 30 s at 95 °C, 1 min at 60 °C, and 1 min at 72 °C, and a final step at 72 °C for 2 min. All PCR products were separated and then directly sequenced using BigDye Terminator v.3.1 Mix (Applied Biosystems, Foster City, CA) and analyzed by capillary electrophoresis using an ABI Prism 3500 Genetic Analyzer (Applied Biosystems).
Multiplex ligation-dependent probe amplification (MLPA)
Deletions or duplications in GLDC,AMT and GCSH genes were analyzed using MLPA [SALSA MLPA P209 Glycine Encephalopathy probe mix (MRC-Holland)]. The analysis was employed according to the manufacturer’s protocol. The MLPA data were analyzed using GeneMarker (version 1.6) software in order to determine potential CN (copy number) variations of exons. The fluorescent signals were compared to normal controls, resulting in a ratio of 0.5 for deletions and 1.5 for duplications. Each test was repeated twice in order to confirm the results.
Quantitative PCR (Q-PCR)
To verify the variants identified by MLPA, Q-PCR was performed by amplifying GLDC exon 3 with two primers sets (Additional file 1: Table S1). The primer sets, referred to as Target 1 and Target 2, were designed to amplify the 3′ and 5′ ends of exon 3 respectively, with the products size being 80-150 bp. The TERT gene was chosen as the endogenous control in this study. All reactions using SYBR Green Dye were run using the following cycle: 2 min at 50 °C, 5 min at 95 °C and 40 cycles of 15 s at 95 °C and 30 s at 60 °C. All reactions were performed using the ABI StepOne real-time PCR system. After Q-PCR was performed, the data was collected and analyzed by the 2-ΔΔCT method. Data using Target 1 and Target 2 primers giving a CN = 1 indicated deletion of exon3 of GLDC gene.
In silico analytical tools
The identified variant was checked for its presence in disease databases such as the Human Gene Mutation Database (HGMD) [10], ClinVar [11] and the Leiden Open Variation Database (LOVD) [12], following which several bioinformatic programs (PolyPhen 2, SIFT, PROVEAN and MutationTaster) were employed to predict the impact of a missense change on the protein structure and function [13,14,15,16] (Additional file 2: Table S2). Additionally, multiple amino acid sequences were extracted from National Center for Biotechnology Information (NCBI) and aligned to verify the evolutionary conservation using ClustalX (http://www.clustal.org/clustal2) [17, 18].
Results
Clinical data and auxiliary examination
Three patients (two males, one female), from one kindred in the Quanzhou area, Fujian Province, southeastern China, were diagnosed with NKH based on their clinical manifestations and an abnormal metabolic profile (Fig. 1a). The non-consanguineous parents are healthy individuals, and their first child was not found to have any clinical symptoms during follow-up. All three affected siblings were born at term with normal birth weight after an uneventful delivery, however, they presented with lethargy, seizures and hypotonia. Both the proband and the younger brother presented with severe clinical symptoms and died during the neonatal period, while the proband’s younger sister, who exhibited relatively milder symptoms, is currently aged 7 months, and has intractable seizures and a profound developmental delay, as shown by magnetic resonance imaging (MRI) (Fig. 1b). Sodium benzoate and dextromethorphan were prescribed for the patient to improve her alertness and decrease seizure frequency. Detailed clinical and biochemical information, as well as the genotypes of the siblings are shown in Table 1.
Mutation identification and bioinformatic analysis
Sanger sequencing identified a novel c.2680A > G variant in exon 23 of GLDC gene in the proband (Fig. 1c). This novel variant has not yet been reported in the literature, and was not found in the 1000 Genome, ESP6500, ExAc or dbSNP databases, and, in addition, could not be detected in 100 healthy individuals. In silico analysis predicted that the novel variant was a deleterious mutation ((Additional file 2: Table S2). Additionally, the Thr residue at position 894 was found to be a highly conserved amino acid residue among different species, as assessed using ClustalX software (Fig. 1f), and so a variant of this residue is likely to be deleterious. MLPA analysis combined with Q-PCR experiments also revealed an exonic deletion event in the proband, namely a heterozygotic deletion of exon 3 (Figs. 2 and 3).

Multiplex ligation-dependent probe amplification analysis of GLDC, AMT and GCSH genes. A heterozygous deletion of exon 3 in GLDC gene was detected in the proband (a) and his father (c), but not in his mother (b)

Copy number variations analysis associates exon 3 of GLDC gene detected by Q-PCR. Two independent primers were used, the results of which are labeled as Target 1 and Target 2 respectively. The proband and his father both having a CN = 1 indicated a heterozygous deletion of exon 3, and his mother had a CN = 2, identical to the control samples, indicating a normal copy numbers of exon 3
These findings indicated that the proband had compound heterozygous variants in GLDC gene, i.e. a c.2680A > G (p.Thr894Ala) variant in exon 23 and a heterozygous deletion of exon 3, which were inherited respectively from the mother and the father (Figs. 1d, e and 2). The same variants were observed in the proband’s younger brother and sister, whereas his unaffected elder brother carried only the maternal variant.
Discussion
Clinically, the patients in this Chinese family are representative of neonatal NKH patients. The proband, along with his younger siblings, exhibited severe and progressive manifestation such as lethargy, hypotonia and seizures, with greatly elevated glycine levels in their plasma and CSF. Molecular genetic analysis by Sanger sequencing and MLPA, identified compound heterozygous variants in GLDC in these three patients, namely a c.2680A > G (p.Thr894Ala) variant in exon 23 and a heterozygous deletion of exon 3, which were inherited respectively from their parents. Although these three siblings have the same compound heterozygous variants in GLDC gene, and similar clinical phenotypes, the severity of the disease, as well as the outcome, were variable. Because of this, we speculate that other genetic and environmental factors may also be responsible for the clinical phenotype including the severity and the outcome, which requires further research.
NKH is primarily caused by mutations in GLDC gene. The 113.15 kb GLDC gene located on chromosome 9p24.1, encodes a 1021 amino acid protein called glycine decarboxylase. To date, more than 122 mutations in GLDC have been reported to cause NKH, including a multitude of missense mutations and different deletions involving multiple GLDC exons [19]. The mutations found in NKH are highly heterogeneous, although recurrent missense mutations reported include the p.R515S mutation found in Caucasians and the p.S564I mutation observed in Finnish populations [6, 20]. Intragenic copy number variations (CNVs) have been noted in approximately 20% of GLDC alleles, the majority of which are multi-exon deletions [21]. These mutated alleles caused by multi-exon deletions or duplications occur in various haplotypes and in different ethnic groups. In China, to the best of our knowledge, only two NKH patients with compound heterozygous mutations in GLDC gene have been reported to date [8, 9].
In this work, we describe the novel variant c.2680A > G, which leads to the substitution of the polar amino acid threonine with the nonpolar animo acid alanine at position 894 of the P-protein. Computational analysis predicted that the variant is likely to have pathogenic significance and a conservation analysis in different species showed that this amino acid was highly conserved across a broad range of species, which again strongly suggests that the variant at this site might be deleterious. The deletion of exon 3 of the GLDC gene is a frameshift variant resulting in the premature termination of the P-protein, and is also likely to be clinically significant. This CNV has previously been reported by Coughlin et al. in a systematic study of 578 families, among which one child had a homozygous deletion of exon 3 [22], and this is the first time this variant has been found in the Chinese population. Although the MLPA technique fails to pinpoint the exact location of this deletion, based on MLPA probes and subsequent qPCR experiments, we know that the upstream breakpoint was from the MLPA probe binding site of exon 2 to qPCR upstream primer 5′ site of exon 3, and the downstream breakpoint was from qPCR downstream primer 3′ site of exon 3 to the MLPA probe binding site of exon 4 (Additional file 3: Figure S1). According to the HGVS nomenclature, the break point can be expressed as NG_016397.1 (NM_000170.2): c. (261_335-95) _ (470 + 111_476) del. In short, in view of the fact that this CNV might be overlooked by conventional sequencing strategies, MLPA or another technique that detects deletions should therefore also be utilized to analyze the presence of potential CNVs in GLDC gene.
Conclusions
In this study, we describe the clinical and genetic features of a Chinese family with three siblings affected with NKH. A novel variant, as well as a previously reported deletion in GLDC gene, were identified in the three affected siblings. Thus, our findings suggest that these two variants in GLDC gene probably underlie the pathogenesis of NKH in this family, and also enrich the mutational spectrum of GLDC gene.
Abbreviations
- CN:
-
Copy number
- CNV:
-
Copy number variation
- CSF:
-
Cerebrospinal fluid
- GCS:
-
Glycine cleavage system
- MLPA:
-
Multiplex ligation-dependent probe amplification
- MRI:
-
Magnetic resonance imaging
- NKH:
-
Non-ketotic hyperglycinemia
- PCR:
-
Polymerase chain reaction
References
Kure S, Kato K, Dinopoulos A, Gail C, DeGrauw TJ, Christodoulou J, Bzduch V, Kalmanchey R, Fekete G, Trojovsky A, et al. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat. 2006;27(4):343–52.
Flusser H, Korman SH, Sato K, Matsubara Y, Galil A, Kure S. Mild glycine encephalopathy (NKH) in a large kindred due to a silent exonic GLDC splice mutation. Neurology. 2005;64(8):1426–30.
Kanekar S, Byler D. Characteristic MRI findings in neonatal nonketotic hyperglycinemia due to sequence changes in GLDC gene encoding the enzyme glycine decarboxylase. Metab Brain Dis. 2013;28(4):717–20.
Bjoraker KJ, Swanson MA, Coughlin CR 2nd, Christodoulou J, Tan ES, Fergeson M, Dyack S, Ahmad A, Friederich MW, Spector EB, et al. Neurodevelopmental outcome and treatment efficacy of benzoate and Dextromethorphan in siblings with attenuated Nonketotic Hyperglycinemia. J Pediatr. 2016;170:234–9.
Bravo-Alonso I, Navarrete R, Arribas-Carreira L, Perona A, Abia D, Couce ML, Garcia-Cazorla A, Morais A, Domingo R, Ramos MA, et al. Nonketotic hyperglycinemia: functional assessment of missense variants in GLDC to understand phenotypes of the disease. Hum Mutat. 2017;38(6):678–91.
Toone JR, Applegarth DA, Coulter-Mackie MB, James ER. Recurrent mutations in P- and T-proteins of the glycine cleavage complex and a novel T-protein mutation (N145I): a strategy for the molecular investigation of patients with nonketotic hyperglycinemia (NKH). Mol Genet Metab. 2001;72(4):322–5.
Van Hove J, Coughlin C II, Scharer G. Glycine encephalopathy. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 2002. 1993-2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1357/.
Gao ZJ, Jiang Q, Chen Q, Xu KM. Clinical and molecular genetic study of nonketotic hyperglycinemia in a Chinese family. Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatrics. 2017;19(3):268–71.
Liu S, Wang Z, Liang J, Chen N, OuYang H, Zeng W, Chen L, Xie X, Jiang J. Two novel mutations in the glycine decarboxylase gene in a boy with classic nonketotic hyperglycinemia: case report. Archivos argentinos de pediatria. 2017;115(4):e225–9.
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–77.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8.
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557–63.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31(13):3497–500.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8.
Chiu CF, Lin JL, Lin JJ, Tseng MH, Lo FS, Chiang MC. Nonketotic Hyperglycinemia of infants in Taiwan. Pediatrics and neonatology. 2016;57(5):420–6.
Kure STM, Kurihara Y, Leisti J, Zalai D, Chuck G, Tada K, Matsubara Y, Narisawa K. Nonketotic hyperglycinemia: mutation spectra of the GLDC and AMT genes in Finnish and non-Finnish populations. Am J Hum Genet. 1999;65:A2406.
Kanno J, Hutchin T, Kamada F, Narisawa A, Aoki Y, Matsubara Y, Kure S. Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. J Med Genet. 2007;44(3):e69.
Coughlin CR 2nd, Swanson MA, Kronquist K, Acquaviva C, Hutchin T, Rodriguez-Pombo P, Vaisanen ML, Spector E, Creadon-Swindell G, Bras-Goldberg AM, et al. The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genetics in medicine : official journal of the American College of Medical Genetics. 2017;19(1):104–11.
Acknowledgements
We thank all the participants for their co-operation.
Funding
No funding to declare
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
YL and WS carried out the mutation analysis and the first draft of the manuscript. ZZ cared for patients and collected the clinical data. QF was mentors who designed and guided the research study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of The Women’s and Children’s Hospital of Quanzhou. The parents of the patients and 100 control subjects signed written informed consent and agree themselves and their children to take part in this study and using the relevant data and information for scientific research.
Consent for publication
We confirm that all the parents of the patients and 100 control subjects signed written informed consent for publication of their own and children’s genetic data, clinical details and/or any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
List of the primers used for Sanger sequencing and Q-PCR (DOCX 13 kb)
Additional file 2: Table S2.
Pathogenicity prediction analysis of GLDC c.2680A > G alteration (DOC 29 kb)
Additional file 3: Figure S1.
The illustration of breakpoints in exon 3 deletion (TIFF 891 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lin, Y., Zheng, Z., Sun, W. et al. A novel compound heterozygous variant identified in GLDC gene in a Chinese family with non-ketotic hyperglycinemia. BMC Med Genet 19, 5 (2018). https://doi.org/10.1186/s12881-017-0517-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0517-1