
Abstract
Background
Alström syndrome is a rare multi-systemic disorder with a broad spectrum of symptoms. This syndrome is characterized by childhood retinal degeneration; sensorineural hearing loss; obesity; type 2 diabetes mellitus; cardiomyopathy; systemic fibrosis; and pulmonary, hepatic, and renal failure.
Case presentation
A Chinese quartet family with two siblings predominantly affected by cone-rod dystrophy and short stature were recruited. The craniofacial dysmorphism and on-set age-of-cone-rod dystrophy in the proband showed a minor intrafamilial variability. Whole genome sequencing was performed to provide the full spectrum of the two siblings’ genetic variations. In this study, we present the patients’ clinical features and our interpretation of the whole genome sequencing data. After examining the data, we focus on two compound heterozygous mutations, (c.3902C > A, p.S1301X; c.6436C > T, p.R2146X) in ALMS1, which are shared by two siblings.
Conclusion
We reported a novel ALMS1 mutation. Whole genome sequencing is a powerful tool to provide the full spectrum of genetic variations for heterogeneous disorders such as Alström syndrome.
Similar content being viewed by others

Background
Alström syndrome (ALMS; MIM# 203800) is a rare multi-systemic autosomal recessive disorder with a broad spectrum of clinical symptoms, including progressive cone-rod dystrophy, sensorineural hearing impairment, truncal obesity, insulin resistance, hypogonadism, dilated or restrictive cardiomyopathy, and multiple organ failure [1, 2]. Alström syndrome is caused by mutations in the ALMS1 gene.
In the past 3 years, several new causative genes for at least 150 heritable disorders, and mutations in known genes for unexplained phenotypes, have been discovered by using the whole-exome sequencing (WES) or whole-genome sequencing (WGS) techniques [3]. Protein-coding genes comprise only approximately 1% of the human genome but harbor 85% of the mutations with substantial effects on disease-related traits [4], which makes WES a cost-effective method for detecting coding-region mutations. Nevertheless, WES involves a hybridization step by which the coding regions are captured from the whole genomic DNA, and during this process, 3 to 5% of the targets will be missed. Furthermore, the full spectrum of copy number variations (CNVs) and breakpoints may not be completely characterized by WES [5]. In contrast to WES, WGS avoids these limitations, and its quartet framework offers an analytical advantage for the detection and correction of sequencing errors [6]. As the price of generating whole-exome and whole-genome data continues to drop and the price difference between WES and WGS continues to narrow, WGS is anticipated to become more widely used in clinical settings [7]. A Chinese quartet family with two siblings primarily affected by cone-rod dystrophy were recruited. To provide for the full spectrum of the patients’ genetic variations, whole-genome sequencing (WGS) was performed to search for candidate point mutations, InDels and CNVs. In this study, we presented the patients’ clinical features and our interpretation of the WGS data. We finally focus on two compound heterozygous mutations in ALMS1 shared by two siblings.
Case presentation
Clinical studies
At the time of admission, the proband was a 16-year-old boy with a height of 155 cm (<3rd centile) (short stature, HP:0004322) and weight of 57 kg (25th–50th percentile) [8]. He had craniofacial dysmorphism (round face HP:0000311, frontal balding HP: 0002292, thin hair HP: 0002213, narrow palpebral fissures HP: 0000581, and bitemporal flatting HP: 0000272, Fig. 1b). There was no evidence of hypponadism at the age of 16 years, with normal testicular volume and normal penis length, with well-developed cavernous. He had a history of photophobia (HP: 0000613) (HP: 0000613) and nystagmus (HP: 0000639) beginning at less than 3 months of age, which eventually resulted in blindness (HP: 0000618) by the age of 7–8 years. Bilateral sensorineural hearing loss (HP: 0000408) developed at the age of 8–9 years. Diabetes (HP: 0000873) developed at the age of 14 and was treated with subcutaneous insulin injections. Obesity developed (HP: 0001513) before 15 years. The ophthalmologic examination was notable for cone-rod dystrophy (HP: 0000548). Abdominal ultrasound found multiple solid masses (5.1–10.3 mm) in the liver. Echocardiography did not indicate any abnormalities at this time. There was no evidence of developmental delays, mental retardation, cardiologic impairment, hypertension, polydactyly, or hypogonadism. Serum concentration levels of triglyceridemia and blood urea nitrogen were slightly elevated, but creatinine, aspartate aminotransferase, alanine, and aminotransferase levels were normal.

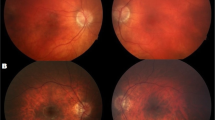
Pedigrees of the family, Sanger sequencing confirmation and Optical coherence tomography of the proband. a Pedigrees of the quartet families. b Optical coherence tomography of the proband reveals atrophy of the retina with epiretinal membrane and retina pigment epithelium irregularly arrangement. c Sanger sequencing confirmation in the quartet family. Heterozygous nucleotide and amino acid substitutions are shown at the top. Presence of the mutated alleles is indicated by a red asterisk
The younger brother is 8 years old, with a height of 120 cm (3rd percentile) and weight 30 kg (50th–75th percentile). There were no obvious malformations such as craniofacial dysmorphism or obesity (BMI = 20.83 kg/m2). There was no evidence of hypponadism at this time. The boy had a history of photophobia, nystagmus, and progressive cone-rod retinal dystrophy beginning at 3 months of age. Vision was 0.1 when evaluated. There was no evidence of hearing loss, diabetes, development delay, or mental retardation. Ophthalmologic examination revealed mild cone-rod dystrophy. Examining abdominal ultrasound, echocardiography, liver function and renal function did not produce evidence of any further abnormalities.
The parents of the two boys were both healthy, non-consanguineous adults. There was no family history of congenital malformations and no known teratogenic exposure. The major clinical manifestations of ALMS are shown in Table 1.
This study was approved by the ethics committee of Children’s Hospital, Fudan University. Informed consent was obtained from the parents, who agreed to join this study, and using the data for scientific research and publication. The methods used in this study were performed in accordance with the approved guidelines.
Whole genome sequencing
This study was approved by the Ethics Committee of Children’s Hospital, Fudan University. Genomic DNA was sequenced on a HiSeq 2000 sequencer according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Clean reads were aligned to the reference human genome (UCSC hg19) using a Burrows-Wheeler Aligner (BWA) (v.0.5.9–r16). The average sequencing depth ranged from 35.73X-38.45 X. The mapping rate of clear data ranged from 97.03 to 97.27%, and the genome coverage ranged from 99.83% to 99.85 (Additional file 1: Table S5).
For CNV detection, we employed a MATLAB packet, SegSeq. Only two de novo CNVs were identified in the proband. However, both CNVs were reported as polymorphisms in the Database of Genomic Variants (DGV).
Single Nucleotide Variants (SNVs) were detected by SOAPsnp (v.1.05). InDels were identified by GATK. ANNOVAR was used to annotate the variants. The number of variants among samples ranged from 6,288,378 to 6,375,627 (Fig. 2a, Additional file 2: Table S1 and Additional file 3: Table S2). Of these variants, there were 1,681,375 to 1,764,264 potential variants of unknown significance (VUS) that did not exist in the dbSNP137 or 1000 Genome Project (Fig. 2b, Additional file 4: Table S3 and Additional file 5: Table S4). After filtering the known variants from the dbSNP137 and 1000 Genome Project and those without potential function effect, there were 803 candidate pathogenic variants in 399 genes shared between the two affected siblings. The similar phenotypes between the two boys, ALMS negative family history, and absence of consanguinity suggest an X-linked or autosomal recessive inheritance. A total of 10 genes matched the heritance model (Additional file 6: Table S6). Then, we found two heterozygous variants in the ALMS1 gene (NM_015120.4, .3902C > A, p.S1301X; c.6436C > T, p.R2146X).

Illustrates the pipeline of whole genome data analysis. a The count distribution of variants (SNVs and InDels) from WGS clean data for each individual in the family. The variants were classified into distinct classes according to the genomic regions. b The count distribution of variants of unknown significance that do not exist in the dbSNP137 and 1000 Genome Project. c the proportion distribution of SNVs and InDels, both of which are shared by two affected siblings. For each group, the variants were divided into different classes according to genomic regions. d the distribution of potentially pathogenic mutations classified by mutation functions
Both ALMS1 mutations were validated by Sanger technique on a 3500XL instrument. S1301X, R2146X are the paternal and maternal variants, respectively. Between the two variants, R2146X was reported most recently in 2015 [9]. S1301X is a novel variant, absent from the Exac, 1000 genome, dbSNP and internal databases: 1554 probands and 1257 families. S1301X, a nonsense mutation located in exon 8, yields a truncated protein missing 2866 amino acids that will likely cause either a partial or complete loss of function. Therefore, we assumed these variants were pathogenic mutations.
Furthermore, different variants between the two siblings were analyzed. A heterozygous missense variant c.1379G > A, p.R460H in BBS1 gene was only identified in the proband. G at position 1379 is a conserved amino acid. The potential pathogenicity of this variant was scored 0.962, 0.01 and 1 by PolyPhen2, SIFT and MutationTaster, respectively; therefore, the variant was classified as being “damaged”.
Discussion
It is difficult to definitively diagnose ALMS solely by the clinical features of the two affected siblings. As described by Marshall et al. [2, 10], diagnostic criteria of Alström syndrome have been revised to take the age of the affected person into consideration (Table 1). In our study, the proband with one major (vision) and two minor (diabetes mellitus and hearing) clinical features do not meet the diagnostic criteria for a 15-year-old child. The 8-year-old brother suffering from vision lesion and short stature with just one major and one minor feature, respectively, fails to meet the criteria for a 3- to 14-year-old child. Furthermore, some clinical phenotypes, such as obesity, male hypogonadism, hepatic dysfunction, proteinuria, chronic bronchitis and developmental delay, were reported to occur within the first decade among 50% of Alström patients. However, neither of the affected siblings developed these abnormalities.
The biological function of the ALMS1 protein remains unclear, although localization to centrosomes and basal bodies of ciliated cells suggest apparent roles in ciliary function, intracellular trafficking and adipocyte differentiation [11]. The complex spectrum of traits for ALMS is consistent with those described for ciliopathies, which are genetic disorders resulting from the alterations of various genes. For example,, Bardet-Biedle syndrome (BBS; MIM# 209900) shares many clinical phenotypes with ALMS: retinitis pigmentosa, deafness, and diabetes mellitus [10, 12]. A total of18 genes, excluding ALMS1, have been associated with BBS [13]. Previous reports suggested that such interactions might extend to cases of non-Mendelian inheritance in the form of triallelism. This defies the prevailing perception of BBS as an autosomal recessive disease [14,15,16]. ALMS is a heterogeneous genetic disease with variable expressivity, even within families.
WGS is a powerful tool that provides the full spectrum of genetic variations for such heterogeneous disorders. WGS can not only detect point mutations and small insertions/deletions (InDels) but may also be a useful technique for identifying large copy number variations and the variants in noncoding regions [5, 6].
In our study, we used WGS data filtering (Fig. 2) to discover two compound heterozygous nonsense mutations in ALMS1 shared by two siblings whose mutations resulted in overlapping clinical presentations between both affected individuals. Thus, we established the diagnosis of ALMS in the two boys.
Previous reports suggested that ciliopathies with a triallelic inheritance defy the prevailing perception among the medical community that BBS is an autosomal recessive disease [17]. For the family in this case study, we identified an extra heterozygous missense variant in the BBS1 gene in the proband only by WGS and speculate that a BBS1 gene variant may interact with ALMS1 to alter the onset and course of manifestations of ALMS. More research is required to confirm these results.
Conclusions
We identified two compound heterozygous ALMS1 mutations in a Chinese quartet family shared between two siblings suffering cone-rod dystrophy and short stature. We report a novel ALMS1 mutation and confirmed that WGS is a powerful tool for providing the full spectrum of genetic variations for such heterogeneous disorders.
Abbreviations
- ALMS:
-
Alström syndrome
- CNVs:
-
Copy number variations
- DGV:
-
Database of genomic variants
- InDels:
-
Insertions/Deletions
- NGS:
-
Next-generation sequencing
- VUS:
-
Variants of unknown significance
- WES:
-
Whole-exome sequencing
- WGS:
-
Whole-genome sequencing
References
Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, Milan G, Zhang W, Wilson DI, Hearn T, et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Hum Mutat. 2007;28(11):1114–23.
Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom syndrome: genetics and clinical overview. Curr Genomics. 2011;12(3):225–35.
Grody WW, Thompson BH, Hudgins L. Whole-exome/genome sequencing and genomics. Pediatrics. 2013;132 Suppl 3:S211–5.
Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–101.
Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC bioinformatics. 2013;14 Suppl 11:S1.
Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90(3):502–10.
Yu Y, Wu BL, Wu J, Shen Y. Exome and whole-genome sequencing as clinical tests: a transformative practice in molecular diagnostics. Clinical chemistry. 2012;58(11):1507–9.
Li H, Ji CY, Zong XN, Zhang YQ. [Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years]. Zhonghua er ke za zhi. 2009;47(7):487–92.
Long PA, Evans JM, Olson TM. Exome sequencing establishes diagnosis of Alstrom syndrome in an infant presenting with non-syndromic dilated cardiomyopathy. Am J Med Genet A. 2015;167A(4):886–90.
Marshall JD, Beck S, Maffei P, Naggert JK. Alstrom syndrome. Eur J Hum Genet. 2007;15(12):1193–202.
Collin GB, Marshall JD, King BL, Milan G, Maffei P, Jagger DJ, Naggert JK. The Alstrom syndrome protein, ALMS1, interacts with alpha-actinin and components of the endosome recycling pathway. PloS one. 2012;7(5):e37925.
Aliferis K, Helle S, Gyapay G, Duchatelet S, Stoetzel C, Mandel JL, Dollfus H. Differentiating Alstrom from Bardet-Biedl syndrome (BBS) using systematic ciliopathy genes sequencing. Ophthalmic Genet. 2012;33(1):18–22.
Scheidecker S, Etard C, Pierce NW, Geoffroy V, Schaefer E, Muller J, Chennen K, Flori E, Pelletier V, Poch O, et al. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J Med Genet. 2014;51(2):132–6.
Beales PL, Badano JL, Ross AJ, Ansley SJ, Hoskins BE, Kirsten B, Mein CA, Froguel P, Scambler PJ, Lewis RA, et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet. 2003;72(5):1187–99.
Laurier V, Stoetzel C, Muller J, Thibault C, Corbani S, Jalkh N, Salem N, Chouery E, Poch O, Licaire S, et al. Pitfalls of homozygosity mapping: an extended consanguineous Bardet-Biedl syndrome family with two mutant genes (BBS2, BBS10), three mutations, but no triallelism. Eur J Hum Genet. 2006;14(11):1195–203.
Abu-Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M, Sirelkhatim MO, Al-Hassnan Z, Alkuraya B, Mohamed JY, et al. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet. 2012;20(4):420–7.
Liu X, Wang ZX, Jin WN, Lv H, Zhang W, Que CL, Huang Y, Yuan Y. Clinical and GAA gene mutation analysis in mainland Chinese patients with late-onset Pompe disease: identifying c.2238G > C as the most common mutation. BMC Med Genet. 2014;15.
Acknowledgments
The authors would like to express their gratitude to the participating family and would also like to thank Prof. Yonghui Jiang of School of Medicine, Duke University for his support and mentorship.
Funding
This work was supported by grants from Shanghai Municipal Commission of Health and Family Planning (2013ZYJB0015) and Science and Technology Commission of Shanghai Municipal (14411950402).
Availability of data and materials
The whole genome sequencing data for each individual are available in the Sequence Read Archive (SRA) under BioProject ID “PRJNA342101”. And the data summary was presented as Additional files within the manuscript.
Authors’ contributions
LY and WZ conceived of this study and provided and participated in its design. LY drafted the manuscript. ZL and WT performed the WGS data analysis and interpretation. XF and MW performed the molecular genetics studies and participated in the sequence alignment. MM performed the follow-up examinations. GZ and HW performed the Sanger confirmation and helped to interpret the data. GH and WZ participated in the design and coordination of this study in addition to revising and critiquing the manuscript. All authors read and approved the final draft of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Informed consent was obtained from the parents, who agreed to join this study, and using the data for scientific research and publication. The methods used in this study were performed in accordance with the approved guidelines.
Ethics approval and consent to participate
This study was approved by the ethics committee of Children’s Hospital, Fudan University. Informed consent was obtained from the parents, who agreed to join this study, and using the data for scientific research and publication.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Summary of the results of the WGS statistics in the family. (PDF 12 kb)
Additional file 2:
Summary of SNV identification in the family. (DOCX 16 kb)
Additional file 3:
Summary of InDels identification in the family. (DOCX 16 kb)
Additional file 4:
Summary of SNV identification after polymorphism in the dbSNP and 1000 Genome Project were filtered. (DOCX 16 kb)
Additional file 5:
Summary of InDels identification after polymorphism in the dbSNP and 1000 Genome Project were filtered. (DOCX 15 kb)
Additional file 6:
Summary of rare and inheritance pattern filtered variants identified in the family. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, L., Li, Z., Mei, M. et al. Whole genome sequencing identifies a novel ALMS1 gene mutation in two Chinese siblings with Alström syndrome. BMC Med Genet 18, 75 (2017). https://doi.org/10.1186/s12881-017-0418-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0418-3