
Abstract
Background
Maturity onset diabetes of the young (MODY) is an autosomal dominant form of non–insulin-dependent diabetes mellitus caused by mutations in at least 13 different genes. The hepatocyte nuclear factor (HNF)-1α gene is affected in the most common form (HNF1A-MODY [MODY3]).
Case presentation
We describe the co-inheritance of a novel heterozygous missense mutation c.1761C > G (p.Pro588Ala) with a novel complex deletion insertion mutation (c.1765_1766delinsGCCCGfs86*) in the HNF-1α gene among affected members of one family. Both mutations were present in the affected patients and neither was present in unaffected family members. The family had not only inheritance of MODY but also increased susceptibility to type 2 diabetes. Therefore one family member had classical type 2 diabetes including metabolic syndrome aggravated by a genetic predisposition in the form of HNF1A-MODY.
Conclusion
The presence of common type 2 diabetes features should not detract from the possibility of MODY in patients with a striking autosomal-dominant family history.
Similar content being viewed by others


Background
Maturity onset diabetes of the young (MODY) comprises a group of clinically and genetically heterogeneous familial disorders with a clinical appearance similar to non–insulin-dependent type 2 diabetes that is caused by mutations in at least 13 different genes (MODY1 to MODY13) [1, 2]. MODY is characterized by autosomal dominant inheritance and early onset diabetes, but otherwise the phenotype appears to be heterogeneous [1–3]. HNF1A-MODY (MODY3) is the most common form of the disorder in white populations and is caused by mutations and, rarely, deletions affecting HNF-1α, a transcription factor that functions in the transcriptional network necessary for liver as well as pancreatic β-cell development and function [4]. HNF1A-MODY is a clinically progressive phenotype with progressive hyperglycemia, glycosuria, and decreased renal function or microvascular complications, which can be reduced substantially by early treatment [1–3, 5]. We report siblings with novel HNF1A-MODY mutations but with different clinical manifestations. This finding may be due to an additional maternal predisposition for type 2 diabetes, which has aggravated the genetic susceptibility burden for the diabetes phenotype in one of the siblings.
Case presentation
The index patient (female, age 43 years at examination) and five further family members agreed to undertake molecular analyses. The proband and three family members were analyzed for serum metabolic parameters; two patients (the index female patient and her male sibling) agreed to ultrasound examination, an interventional oral glucose tolerance test (oGTT) and lipid tolerance testing. For ethical reasons family members age <18 years and individuals with further overt clinical symptoms did not undergo interventional investigations. All interventions were performed at the Asklepios Clinik St. Georg (Hamburg, Germany). Written informed consent was obtained from all participants (parents provided informed consent on behalf of subjects age <18 years). The study protocol and procedure were approved by the parents of the participating children and the ethical committee of the University of Hamburg. The study was conducted according to principles laid out in the Declaration of Helsinki.
Determination of metabolic parameters
Blood and serum samples were collected according to standard protocols. Automated standard systems for measuring clinical variables, including insulin, glucose, hemoglobin A1c, cholesterol (including high-density and low-density lipoprotein) and triglycerides, were used. Standard surrogate indexes were calculated from fasting blood glucose and plasma insulin concentrations as follows: quantitative insulin sensitivity check index QUICKI = 1/(log(I0) + log(G0)), where I0 is fasting insulin (μU/ml) and G0 is fasting glucose (mg/dl); and homeostatic model assessment of insulin resistance HOMA-IR = (G0 * I0)/22.5, with fasting glucose expressed as mmol/l and fasting insulin expressed as μU/ml. Creatinine, albumin and glucose concentrations were determined from urine samples. To exclude the presence of type 1 diabetes or latent autoimmune diabetes of adults, measurement of antibodies against insulin, GAD, thyrosine phosphatase (IA2), and islet cells (ICA) were determined. Blood pressure, ophthalmoscopy and angiography of lower extremities were determined in 3 probands.
Molecular analyses
Genomic DNA was extracted from whole blood using the blood extraction kit (Qiagen, Hilden, Germany). The targeted resequencing approach of glucokinase, HNF-1α, HNF-1β, HNF-4α, IPF-1/PDX-1 and NeuroD1/β2 including all splice variants and related promoter areas corresponding to the molecular basis of the most prevalent MODY1 to MODY6 have been reported [6]. Nucleotide numbering of HNF-1α was given according to NM_000545.5.
Metabolic testing
To determine fat tolerance, a breakfast with a total energy content of 1080 kcal (47 % fat, 40 % carbohydrates and 13 % proteins) was served after a 10-h fasting period under current medication. Blood was drawn before the meal (0 min) and 120 min and 240 min after food consumption [7]. To test oGTT, blood samples were drawn before (0 min) and 60 min and 120 min after a 75 mg oral glucose load under current antidiabetic therapy.
Results
The index patient reported early onset of diabetes at age 25 years. No type 1 diabetes specific antibody pattern was detected, but proteinuria and recurrent nephrolithiasis were reported. In the index patient, diabetes had been diagnosed 3 years before the first pregnancy and she developed obstetric diabetic complications during all of her four pregnancies, which were treated with insulin. Two pregnancies were successful, but both newborns developed reversible jaundice, macrosomia and hypoglycemia within the first 2 weeks.
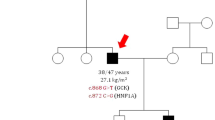
Examination of the family history revealed that her brother also showed early onset diabetes (at 16 years), whereas her parents were both diagnosed as diabetic in routine health screens. The father was diagnosed at the age of 38 years without microvascular complications, including a stable HbA1c of <7.0 % whereas the mother had typical late onset diabetes diagnosed at age 56 years. Thus the family history indicated type 2 diabetes at the maternal site and diabetes with autosomal heritage and early onset at the paternal site. The positive family history, especially with clinical manifestation at a young age with lack of diabetes-related autoantibodies, is a feature typical of MODY. In accordance with this finding, we identified a novel heterozygous missense mutation c.1761C > G (p.Pro588Ala) in the index patient. Moreover, a novel complex deletion insertion mutation at c.1765_1766delinsGCCCGfs86*, resulting in a frameshift in exon 9 of the HNF-1α gene (Fig. 1a), was detected. Both mutations were inherited together and co-segregated with an early onset diabetes phenotype in the family, indicating the genotype–phenotype association over three generations. One child of the family was a carrier of the mutations and diagnosed diabetic at the age of 12 with blood glucose 147 mg/dl and HbA1c 6,9 % (Fig. 1b).

Co-segregation of the mutations and key clinical characteristics. a A heterozygous mutation (c.1761C > G (p.Pro588Ala)) and a complex deletion insertion mutation (c.1765_1766delinsGCCCGfs86*) in exon 9 of the HNF-1α gene were identified by direct sequencing (upper panel: control, middle patient forward strand, below: patient reverse strand). Horizontal arrows indicate direction of sequencing; vertical arrows indicate sequence alterations. b Co-segregation of the HNF-1α mutations in the family. The genetic status, age of onset of diabetes, current therapy and date of birth are indicated. Numbers indicate the index patient and the brother. (Black symbol: early onset diabetes phenotype; grey symbol: late onset type 2 diabetes NM: heterozygous mutation present; NN: no mutation; INS: insulin; OHA: oral hypoglycemic agents; SU: sulfonylurea). In detail, at time of investigation the medication was as follows: index patient (insulin, angiotensin-converting-enzyme inhibitors), brother (glinide), daughter (metformin), father (insulin, statin, glibenclamide), and mother (metformin, glibenclamide, statin, angiotensin-converting-enzyme inhibitors)
Although the mutations were present in both overweight siblings, their metabolic phenotypes differed (Table 1). The index patient (body mass index 27 kg/m2) showed elevated fasting glucose and insulin concentrations, increased HbA1c, moderately elevated triglyceride concentrations and dyslipidemia. Ophthalmoscopy, angiography of the lower extremities and ultrasound examination of the abdomen were performed. No microvascular complications were identified, but low-grade hepatic steatosis and an unaffected pancreas were determined. The brother also showed elevated fasting blood glucose and proteinuria, but no type 1 diabetes-specific antibodies or microvascular complications were detectable. In contrast to the index patient, fasting insulin was normal and HbA1c was significantly increased. Furthermore, features of the metabolic syndrome, including obesity (body mass index 30 kg/m2) and dyslipidemia, were more pronounced. Ultrasound examination indicated severe homogenous steatosis and liver enlargement with low-grade splenomegaly and a lipomatous pancreas. The patient was also a tobacco smoker, a further risk factor.
To further characterize potential differences in the metabolic response, the siblings were challenged with oral glucose and separately with a lipid-rich meal (Tables 2 and 3). The index patient showed a diabetes-specific pattern in all variables analyzed in the oGTT. The brother was in the near-normal range except for his triglyceride concentration. As the fasting triglyceride concentration was pathologically high, the values showed only an average decrease to 4.5 mmol/l under glucose load. The lipid profile was aggravated under lipid tolerance tests. Following lipid load, glucose concentrations in the siblings are nearly identical but were elevated compared with the normal range [8–10]. Most strikingly, in the brother, during the lipid tolerance test fasting triglycerides increased from 8.5 mmol/l to >11.4 mmol/l and declined to 10.1 mmol/l within 2 h. Again, the vast increase in triglyceride concentrations in the lipid tolerance test was not observed in the index patient.
Conclusions
We identified two sequence variations in the HNF-1α gene in siblings with divergent clinical manifestations of type 2 diabetes, metabolic syndrome and HNF1A-MODY mutations. One is a heterozygous mutation of c.1761C > G (p.Pro588Ala). The other is a complex deletion insertion mutation at c.1765_1766delinsGCCCGfs86* altering p.Thr589Ser and inserting a further p.P589 + 1 in exon 9 but not affecting the splicing site directly. The mutations identified were not reported in the exome sequencing-based database of human variation consisting of approximately 60,000 individual samples (http://exac.broadinstitute.org/gene/ENSG00000135100). A mutation of 1785C > T (p.Pro588Ser) has been reported in a previous study of MODY families [11], and Colclough et al. [12] observed a frameshift deletion mutation c.1765_1766delCA at the identical site of the mutation identified here. Although both previous reports did not describe the affected families [11, 12], these observations indicate the importance of this area of the HNF-1α gene as a mutation hotspot. This, alongside the dominant inheritance and the co-segregation with diabetes phenotype, stresses the hypothesis that the mutation causes the MODY phenotype in the family described here.
HNF-1α is composed of three functional domains, and three isoforms are generated by alternative splicing, with different transcriptional properties and tissue expression patterns [13, 14]. Mechanistically both mutations affect the transactivation domain of the HNF-1α isoform A with the isoform specific exons 8 to 10 of the transactivation domain, which is mainly expressed in the fetal pancreas [14]. An older age at onset was observed in HNF1A-MODY patients carrying a HNF-1α missense mutation affecting specifically the HNF-1α (A) isoform (10B) [11]. Interestingly, in vitro studies indicated that truncation of exon 10 decreases transcriptional activation by HNF-1α by approximately 50 % [15]. Although the splicing site is not affected directly, as a functional consequence after splicing the reading frame of the entire exon 10 is shifted to generate an 85 AA sequence without any homology to known protein sequences. As the HNF-1α isoform A is mainly expressed in the fetal pancreas [14], one may speculate that identified mutations interfere with susceptibility to type 2 diabetes on the level of pancreas functionality or maturation.
MODY in adults is often clinically misdiagnosed as type 1 or type 2 diabetes [3]. Therefore it is tempting to speculate that HNF1A-MODY is under-diagnosed in the ‘sink’ of common type 2 diabetes. HNF1A-MODY is a clinically progressive phenotype with hyperglycemia, glycosuria, and decreased renal function or microvascular complications, and usual associations with microvascular and macrovascular complications commensurate with glycemic control [1, 2, 16]. In our family, HNF1A-MODY presents clinically as diabetes without dyslipidemia, as reflected by the index patient. In contrast, HNF1A-MODY was aggravated in the brother by classical insulin resistant type 2 diabetes or features of metabolic syndrome indicated by dyslipidemia with severe hypertriglyceridemia and hepatic steatosis.
Family history and triglyceride concentrations are of special importance in the differentiation of insulin-resistant type 2 diabetes and HNF1A-MODY. In general, type 2 diabetes is characterized by significantly higher triglyceride values and lower high-density lipoprotein cholesterol compared with HNF1A-MODY or non-diabetics, with a high-density lipoprotein cholesterol > 1.12 mmol/l discriminates HNF1A-MODY from type 2 diabetes with 75 % sensitivity and 64 % specificity [17].
On the other hand increased circulating serum lipids are also a combinatory risk factor to various metabolic diseases such as type 2 diabetes. To investigate whether circulating lipids are of special importance in such combined diabetes types, we challenged the affected siblings with lipid and glucose tolerance tests, but gained no further insight beside the present basal clinical features of type 2 diabetes or a more severe insulin-resistant phenotype in the affected brother. Therefore these investigations did not shed light on further therapeutic approaches except therapy for type 2 diabetes.
In our patients, one possible hypothesis for the differences in lipid metabolism observed might be the differing distribution of the maternal genetic predisposition for type 2 diabetes (e.g. in heterozygous states). In combination with the genetic predisposition of the HNF1A-MODY-relevant mutations, this resulted in an exponential metabolic risk, which may be triggered by lifestyle factors. Although in cases with occurrence of type 2 diabetes and HNF1A-MODY-relevant mutations such as this, treatment of type 2 diabetes is the major clinical aim, therapy should be optimized according to the patients’ genetic profile because a MODY-relevant mutation alters the therapeutic needs. Mutations affecting HNF-1α transcriptional activity interfere with activation of cellular HNF-1α targets, which has been shown to decrease the uptake of sulfonylurea, resulting in increased circulating levels [18]. Therefore patients with HNF1A-MODY have been shown to be hypersensitive to sulfonylurea therapy [19, 20], which makes sulfonylurea the therapy of choice. Nevertheless, tight dose control may be needed for the individual patient. Taken together, the presence of common type 2 diabetes features should not detract from the possibility of MODY in patients with a striking autosomal dominant family history, in order to optimize the individual therapy.
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
References
Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 2011;34(8):1878–84. doi:10.2337/dc11-0035.
McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem. 2013;50(Pt 5):403–15. doi:10.1177/0004563213483458.
Thanabalasingham G, Pal A, Selwood MP, Dudley C, Fisher K, Bingley PJ, et al. Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care. 2012;35(6):1206–12. doi:10.2337/dc11-1243.
Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303(5662):1378–81. doi:10.1126/science.1089769.
Bellanne-Chantelot C, Levy DJ, Carette C, Saint-Martin C, Riveline JP, Larger E, et al. Clinical characteristics and diagnostic criteria of maturity-onset diabetes of the young (MODY) due to molecular anomalies of the HNF1A gene. J Clin Endocrinol Metab. 2011;96(8):E1346–51. doi:10.1210/jc.2011-0268.
Knebel B, Jacob S, Boxberg CV, Muller-Wieland D, Kotzka J. A novel nonsense mutation in GCK exon 9 co-segregates with diabetes phenotype. Exp Clin Endocrinol Diabetes. 2004;112(6):298–301. doi:10.1055/s-2004-820967.
Vossen M, Todter K, Altenburg C, Beisiegel U, Scheja L. Plasma triglycerides after oral glucose load specifically associate with metabolic risk markers in healthy type 2 diabetes offspring. Atherosclerosis. 2011;217(1):214–9. doi:10.1016/j.atherosclerosis.2011.03.013.
Musso G, Cassader M, De Michieli F, Rosina F, Orlandi F, Gambino R. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology. 2012;56(3):933–42. doi:10.1002/hep.25739.
Koutsari C, Malkova D, Hardman AE. Postprandial lipemia after short-term variation in dietary fat and carbohydrate. Metabolism. 2000;49(9):1150–5. doi:10.1053/meta.2000.8612.
de Ugarte MT, Portal VL, Dias AA, Schaan BD. Metabolic response to oral lipid overload in diabetes and impaired glucose tolerance. Diabetes Res Clin Pract. 2005;69(1):36–43. doi:10.1016/j.diabres.2004.11.011.
Bellanne-Chantelot C, Carette C, Riveline JP, Valero R, Gautier JF, Larger E, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2008;57(2):503–8. doi:10.2337/db07-0859.
Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 2013;34(5):669–85. doi:10.1002/humu.22279.
Bach I, Mattei MG, Cereghini S, Yaniv M. Two members of an HNF1 homeoprotein family are expressed in human liver. Nucleic Acids Res. 1991;19(13):3553–9.
Harries LW, Ellard S, Stride A, Morgan NG, Hattersley AT. Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum Mol Genet. 2006;15(14):2216–24. doi:10.1093/hmg/ddl147.
Bjorkhaug L, Bratland A, Njolstad PR, Molven A. Functional dissection of the HNF-1alpha transcription factor: a study on nuclear localization and transcriptional activation. DNA Cell Biol. 2005;24(11):661–9. doi:10.1089/dna.2005.24.661.
Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med. 2010;27(2):157–61. doi:10.1111/j.1464-5491.2009.02913.x.
McDonald TJ, McEneny J, Pearson ER, Thanabalasingham G, Szopa M, Shields BM, et al. Lipoprotein composition in HNF1A-MODY: differentiating between HNF1A-MODY and type 2 diabetes. Clin Chim Acta. 2012;413(9–10):927–32. doi:10.1016/j.cca.2012.02.005.
Boileau P, Wolfrum C, Shih DQ, Yang TA, Wolkoff AW, Stoffel M. Decreased glibenclamide uptake in hepatocytes of hepatocyte nuclear factor-1alpha-deficient mice: a mechanism for hypersensitivity to sulfonylurea therapy in patients with maturity-onset diabetes of the young, type 3 (MODY3). Diabetes. 2002;51 Suppl 3:S343–8.
Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med. 2009;26(4):437–41. doi:10.1111/j.1464-5491.2009.02690.x.
Kleinberger JW, Pollin TI. Personalized medicine in diabetes mellitus: current opportunities and future prospects. Ann N Y Acad Sci. 2015;1346(1):45–56. doi:10.1111/nyas.12757.
Acknowledgments
We are grateful to the members of the family involved in this study. Sophie Rushton-Smith (Medlink Healthcare Communications) provided editorial services on behalf of the authors. The publication of this article was funded by the Open Access fund of the Leibniz Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors confirmed they have contributed to this paper. BK and JK performed molecular analyses, evaluated the data and wrote the manuscript. SM, JH and MKH-F performed specialized medical examinations of the family. OS is the referring physician. DM-W coordinated the study, evaluated the data and wrote the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Knebel, B., Mack, S., Haas, J. et al. Divergent phenotypes in siblings with identical novel mutations in the HNF-1α gene leading to maturity onset diabetes of the young type 3. BMC Med Genet 17, 36 (2016). https://doi.org/10.1186/s12881-016-0297-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-016-0297-z