
Abstract
Background
We report on two brothers with a distinct syndromic phenotype and explore the potential pathogenic cause.
Methods
Cytogenetic tests and exome sequencing were performed on the two brothers and their parents. Variants detected by exome sequencing were validated by Sanger sequencing.
Results
The main phenotype of the two brothers included congenital language disorder, growth retardation, intellectual disability, difficulty in standing and walking, and urinary and fecal incontinence. To the best of our knowledge, no similar phenotype has been reported previously. No abnormalities were detected by G-banding chromosome analysis or array comparative genomic hybridization. However, exome sequencing revealed novel mutations in the ATP-binding cassette, sub-family D member 1 (ABCD1) and Dachshund homolog 2 (DACH2) genes in both brothers. The ABCD1 mutation was a missense mutation c.1126G > C in exon 3 leading to a p.E376Q substitution. The DACH2 mutation was also a missense mutation c.1069A > T in exon 6, leading to a p.S357C substitution. The mother was an asymptomatic heterozygous carrier. Plasma levels of very-long-chain fatty acids were increased in both brothers, suggesting a diagnosis of adrenoleukodystrophy (ALD); however, their phenotype was not compatible with any reported forms of ALD. DACH2 plays an important role in the regulation of brain and limb development, suggesting that this mutation may be involved in the phenotype of the two brothers.
Conclusion
The distinct phenotype demonstrated by these two brothers might represent a new form of ALD or a new syndrome. The combination of mutations in ABCD1 and DACH2 provides a plausible mechanism for this phenotype.
Similar content being viewed by others

Background
Adrenoleukodystrophy (ALD; OMIM#300100) is a serious progressive, genetic disorder that affects the adrenal glands, the spinal cord and the white matter of the nervous system. It is thought to be caused by genetic defects in the ATP-binding cassette, sub-family D member 1 (ABCD1) (OMIM*300371) gene [1],[2]. ALD is characterized by variations in phenotypic expression; seven ALD forms have been reported so far, including childhood cerebral, adolescent cerebral, adult cerebral, adrenomyeloneuropathy (AMN), Addison-only, asymptomatic or presymptomatic, and olivo-ponto-cerebellar ALD [3],[4]. However, there is no exact genotype-phenotype correlation between ABCD1 defects and clinical phenotype. Furthermore, the combination of defects in ABCD1 with defects in other genes, such as the hemophilia A gene [5] or DXS1357E[6], can complicate and worsen the clinical phenotype.
We present two brothers with a distinct phenotype including congenital language disorder, growth retardation, severe intellectual disability, inattention, dysphoria, drooling, heterophony, difficulty in walking and standing without aid, standing on tiptoe with aid, urinary incontinence, and fecal incontinence. To the best of our knowledge, no cases with a similar phenotype have been reported previously. We performed cytogenetic tests and exome sequencing in the two brothers and their parents to screen for potential pathogenic causes.
Methods
Subjects
Two brothers and their parents, as well as 500 unaffected individuals (250 males and 250 females) of matched geographical ancestry, were enrolled in this study. The study was approved by the Shenzhen People's Hospital Ethics Committee, which abides by the Helsinki Declaration on ethical principles for medical research involving human subjects. Written informed consent was obtained from all the participants or their guardians.
DNA extraction
Genomic DNA was obtained from peripheral blood lymphocytes from all individuals, using standard procedures [7].
Cytogenetic analysis
G-banding chromosome analysis (~850 bands) was performed on cultures of peripheral blood lymphocytes from the two brothers and their parents, according to standard techniques [7].
Array comparative genomic hybridization
Array comparative genomic hybridization (array-CGH) was performed using Agilent Technologies' Array CGH Kits (Santa Clara, CA, USA). This platform is 60-mer oligonucleotide-based microarray that allows genome-wide survey and molecular profiling of genomic aberrations with a resolution of ~20 kb (Kit 244A). DNAs were labeled by random priming (Agilent Technologies) for 2 h using Cy5-dUTP for test DNAs and Cy3-dUTP for reference DNAs. Labeled products were column-purified. After probe denaturation and pre-annealing with 50 μg of Cot-1 DNA, hybridization was performed at 65°C with rotation for 40 h. After two washing steps, the arrays were analyzed using an Agilent scanner and Feature Extraction 10.5.0.1 software. The data were analyzed using CGH Analytics 4.0 software (Agilent Technologies). The Aberration Detection Method 2 algorithm was used to identify aberrant intervals.
Exon capture and sequencing, read mapping and single nucleotide polymorphism detection
Targeted capture and massive parallel sequencing of approximately 201,904 coding exons from genomic DNA from the two patients and their mother were performed using the Agilent SureSelect Human All Exon kit, following the manufacturer's protocols. Briefly, genomic DNA was sheared by sonication and the DNA fragments were then purified using a QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). The fragment ends were repaired and adaptors were ligated to the fragments (NEBNext DNA sample prep, New England Biolabs). The adapter-ligated templates were purified using Agencourt AMPure SPRI beads and fragments with an insert size of about 250 bp were excised. Extracted DNA was amplified by ligation-mediated polymerase chain reaction, purified, and hybridized to the SureSelect Biotinylated RNA Library `baits' (Agilent) for enrichment. Each captured library was then loaded on a HiSeq 2000 platform for sequencing. Raw image files were processed using Illumina Pipeline (v1.6) for base-calling with default parameters. SOAPaligner (v2.01) was used to align the sequencing reads to the NCBI human genome reference assembly (build 36.3). Reads that aligned to the designed target region were collected for single nucleotide polymorphism (SNP) identification and subsequent analysis. The consensus sequence and quality of each allele was calculated by SOAPsnp.
Detection of insertions and deletions
Insertions and deletions (Indels) in the exome regions were identified by de novo assembly of the sequencing reads. The reads were assembled using SOAPdenovo with the 31-mer option enabled and the assembled consensus sequences were then aligned to the reference genome by LASTZ. The alignment result was passed to axtBest to separate orthologous from paralogous alignments. Finally, breakpoints in the alignment were identified and the genotypes of Indels were annotated.
Variant analysis
To distinguish between potentially pathogenic mutations and other variants, we only focused on non-synonymous (NS) variants, splice acceptor and donor site mutations (SS), and short coding Indels, anticipating that synonymous variants would less likely to be pathogenic. The variants were compared and filtered using public databases, including dbSNP (v129), 1000 Genome Project (20100208 release), eight HapMap exomes, and YH genome. A novel variant was defined as one that did not exist in these datasets. Only recessive models of inheritance (autosomal recessive model and X-linked recessive model) were considered because of the normal phenotypes of the parents.
Mutation validation
Variants detected by exome sequencing were validated by Sanger sequencing.
Results
Clinical findings
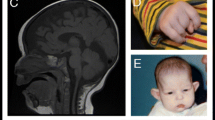
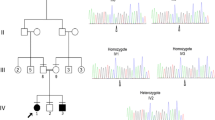
Patient 1: A 10-year-old boy was the result of the third pregnancy of a 33-year-old, gravida six, para three, Chinese woman. The father was 34 years old. The parents were non-consanguineous and healthy. The pregnancy was uncomplicated and the mother denied any exposure to alcohol, teratogenic agents, irradiation or infectious diseases during the pregnancy. The boy was born at full-term and delivered by spontaneous vaginal delivery. His birthweight was 2,500 g (3rd-10th centile), length was 46°Cm (<3rd centile), and head circumference was 30°Cm (<3rd centile). His growth continued to be retarded, and his weight was 28.5 kg (<50th centile), length was 101°Cm (<3rd centile), and head circumference was 46.5°Cm (<3rd centile) at 8 years old. He was noted to have global developmental delay, particularly affecting expressive language. He had never spoken using phrases or `word combinations'. His hearing and vision were normal. He had severe intellectual disability (IQ < 25) and response retardation. His cognition, attention and memory were impaired. He was often restless, crying and drooling, was interested in strangers or novel things, and was frequently incontinent. He had dental dysplasia, with aberrant, dust-colored teeth; the maxillary teeth 11 and 21 were reversed, the 21 and 22 dental crowns were incompletely erupted, bilateral deciduous canines were ablated, and the distal ends of both maxillary and mandibular permanent dental crowns were moderately or mildly tilted (Figure 1). His motor skills were impaired: his manual dexterity and finger tapping were poor, he was able to pick up chopsticks or a spoon, but could not pick up small items or thread a needle. He was never able to walk or stand without aid, and with aid, was only able to achieve a wobbling walk on tiptoe (Figure 2). He was unable to raise or flex his lower limbs freely, and they were ice-cold and insensitive to pain, heat and cold, leading to frequent frostbite and burning (Figure 3). Laboratory tests, including white blood cell and platelets counts, bleeding time, sedimentation rate, serum electrolytes and protein, blood glucose, immune electrophoresis, C-reactive protein, C3 complement, autoantibody screening, and antistreptolysin titer revealed no pathologic findings. The concentrations of cortisol and adrenocorticotropic hormone (ACTH) were normal, though most sex hormones were lower than the reference values (Table 1). The plasma level of very-long-chain fatty acids (VLCFA) was elevated, with a C24/C22 ratio of 1.342 (reference interval: 0.788-1.090), and C26/C22 ratio of 0.086 (reference interval: 0.018-0.038). Sonography, X-ray examination, magnetic resonance imaging (MRI) and computed tomography (CT) scan showed no obvious abnormalities. The mother had previously aborted three times and her first child died of liver cancer at the age of 6 years. There was no family history of chromosomal anomaly or similar case, until the birth of a younger brother (patient 2) (Figure 4).

The poorly developed permanent teeth of the patient 1.

Only with the aid, the patient 1 was able to stand on tiptoe.

The lower limbs of the patient 1 showing many wounds caused by frostbite or burn unconsciously.

The Pedigree of the family. The open square: normal male individual; the closed square: affected male individual; the closed square with an arrow: male proband; the open square with a slash: deceased male individual; the small closed circle: abortion; the open large circle with a closed circle in center: asymptomatic female carrier.
Patient 2: The younger brother of patient 1 was born as the sixth child, 6 years after patient 1, at 40 weeks of gestation after an uncomplicated pregnancy and delivery. His birthweight was 2500 g (3rd-10th centile), length was 45°Cm (<3rd centile), and head circumference was 31°Cm (3rd-10th centile). He started learning to walk at 1 year old, but his lower limbs were weak. He failed to thrive, and his weight was 9 kg (<3rd centile), length was 80.5°Cm (<3rd centile), and head circumference was 42.5°Cm (<3rd centile) at 2.5 years. He had severe intellectual disability (IQ < 25) and response retardation. He was inattentive and often drooling, restless and crying. The muscular tension in his lower limbs was high, and his muscle strength was low. He was unable to stand, walk or speak, and was only able to stand with assistance at the age of 3 years (Figure 5). He was frequently incontinent. Sonography, X-ray examination, MRI and CT scan showed no obvious abnormalities. Auditory brainstem response examination revealed a high frequency hearing threshold in both ears (left ear: 60 db, right ear: 105 db). The concentration of ACTH was normal, but cortisol and sex hormones were low (Table 1). The plasma level of VLCFA showed an increased C24/C22 ratio of 1.428 (reference interval: 0.788-1.090), and C26/C22 ratio of 0.092 (reference interval: 0.018-0.038). No obvious abnormalities were found by other laboratory tests.

The patient 2 was only able to stand with assistance.
Genetic findings
The karyotypes of the two brothers and their parents were normal according to G-banding chromosome analysis. No submicroscopic chromosome aberrations were detected by array-CGH.
The exomes in the two brothers and their mother were sequenced. A total of 35,495,497 bases in 244,069 exons from 21,326 genes were targeted for sequencing. An average of 3.8 Gb of effective sequence was generated per individual as 90 bp paired-end reads; 2.9 Gb (76.3%) passed the quality assessment and aligned to the human reference sequence, and 63.3% of the total bases mapped to the targeted bases with a mean coverage of 64.3-fold (Table 2). About 96% of targeted bases were sufficiently covered to pass our threshold for variant calling. After identification of variants, we focused on NS, SS and Indels, which were more likely to be pathogenic mutations than other variants. Each sibling had one or more NS/SS/Indel variants in ~3,000 genes. Using public databases (dbSNP129, 1000 Genomes Project, eight HapMap exomes and YH genome) as filters, we identified 51 and 33 novel mutated genes in the two brothers, respectively. We compared these genes from the two brothers with their mother to exclude variants that were not compatible with an autosomal recessive model or X-linked recessive model. This reduced the number of candidate genes to 14 and eight, respectively. Three candidate genes were shared by both brothers: one autosomal gene GNAQ (OMIM*600998) and two X chromosomal genes ABCD1 and DACH2 (OMIM*300608) (Table 3).
Sanger sequencing demonstrated that the GNAQ mutations were false positives, but the mutations in DACH2 and ABCD1 were accurate. The ABCD1 mutation was a missense mutation c.1126G > C in exon 3, leading to a p.E376Q substitution (Figure 6), and the DACH2 mutation was also a missense mutation c.1069A > Tc.1069A > T in exon 6, leading to ap.S357C substitution (Figure 7). The mother was an asymptomatic heterozygous carrier (Table 4). Sanger sequencing of 500 unaffected individuals of matched geographical ancestry failed to detect the ABCD1 mutation, while the DACH2 mutation was present in one female. BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) indicated that both the mutations, c.1126G > C transition (E376Q) of ABCD1 and c.1069A > T transition (p.S357C) of DACH2, occurred in highly conserved positions (Figure 6, 7). To assess the likelihood that the two variants had functional impacts on the respective proteins, the biophysical consequences of these variants were predicted using PolyPhen-2. The variant c.1126G > C transition (E376Q) of ABCD1 was considered likely to be functionally benign, while the variant c.1069A > T transition (p.S357C) of DACH2 was likely to be functionally damaging.

Genomic structure of the exons encoding the open reading frame of ABCD1 and identified mutations. ABCD1 is composed of ten exons that encode untranslated regions (UTR) (orange) and protein coding sequence (blue) (upper panel). Sanger sequence of codons 375-377 in exon 3 of ABCD1 indicated the same mutation was present in two brothers and their mother (middle panel). E376Q missense mutation was at a highly conserved position in ABCD1 shown by comparison to the corresponding sequence of six vertebrates (lower panel). Rattus = Rattus norvegicus; Cricetulus = Cricetulus griseus; Pan = Pan troglodytes; Canis = Canis lupus familiaris; Bos = Bos Taurus.

Genomic structure of the exons encoding the open reading frame of DACH2 and identified mutations. DACH2 is composed of twelve exons that encode untranslated regions (UTR) (orange) and protein coding sequence (blue) (upper panel). Sanger sequence of codons 356-358 in exon 6 of DACH2 indicated the same mutation was present in two brothers and their mother (middle panel). S357C missense mutation was at a highly conserved position in DACH2 shown by comparison to the corresponding sequence of six vertebrates (lower panel). Rattus = Rattus norvegicus; Cricetulus = Cricetulus griseus; Pan = Pan troglodytes; Canis = Canis lupus familiaris; Bos = Bos Taurus.
Discussion
We report on two brothers who presented with a distinct phenotype. The potential pathogenic cause was investigated using G-banding chromosome analysis and high-resolution array-CGH in the two brothers and their parents, but no abnormalities were detected. Exome sequencing was therefore performed on the brothers and their mother, to detect genetic variations. The small sample and uncertain pathogenesis of the disease meant that both autosomal recessive and X-linked recessive models were possible. The rarity of the disorder makes it unlikely that causative variants would be present in the general population, and we therefore compared our detected variants with those in dbSNP129, 1000 Genomes Project, eight HapMap exomes and YH genome to eliminate shared variants. Sequencing and comparison of the coding region from the affected brothers and their unaffected mother, and filtering out of the benign changes using public databases, led to the identification of three candidate genes: one autosomal gene GNAQ and two X chromosomal genes ABCD1 and DACH2. Subsequent Sanger sequencing showed that the mutations in GNAQ were false positives.
The ABCD1 mutation was a novel missense mutation, c.1126G > C transition (E376Q), in exon 3. Several lines of evidence support a causative role for this mutation in the brothers' phenotype: 1) ABCD1 mutation is consistent with an X-linked recessive model, which was one of the two possible inherited models; 2) the ABCD1 mutation is not present in the public databases, including dbSNP129, 1000 Genomes Project, eight HapMap exomes and YH genome, and was not found in 500 normal ethnicity-matched controls, excluding the possibility of an amino acid substitution polymorphism; and 3) comparative analyses of ABCD1 in other species show that E376 is conserved among primates, rodents, and other vertebrate species.
ABCD1 encodes an integral peroxisomal membrane protein (ALD protein) that belongs to the ATP-binding cassette-transporter superfamily [6]. The peroxisomal ATP-binding cassette transporter is involved in the import of VLCFA into the peroxisome. Defects in ABCD1 have been shown to be associated with impaired peroxisomalociated with impaired peroxisomal β-oxidation and accumulation of saturated VLCFA in all tissues of the body, and are considered to be the underlying cause of ALD [1],[3].
ALD is a rare X-linked demyelinating disorder affecting the nervous system, adrenal cortex and testis [1],[2], and is characterized by variation in phenotypic expression [6],[8],[9]. To date, several clinical forms have been reported in male patients (Table 5) [3],[4],[10]-[12]. The cerebral forms, including childhood cerebral, adolescent cerebral and adult cerebral, are associated with an inflammatory reaction in the cerebral white matter and progressive neurological damage with rapid evolution, leading to a vegetative state within 6 months to 2 years after onset of symptoms, followed by death at variable ages [13],[14]. In contrast, AMN mainly involves the spinal cord and peripheral nerves, the inflammatory response is absent or mild, and its progression is slower. AMN usually presents with initial symptoms in adult men aged from 20 years to middle-age. Affected individuals develop progressive stiffness and weakness in the legs, sphincter control abnormalities, and sexual dysfunction, and may also have serious cognitive and behavioral disturbances over the decades [12]. Addison-only individuals present with signs of adrenal insufficiency between 2 years of age and adulthood; the signs include unexplained vomiting, weakness or coma. Individuals with asymptomatic ALD usually have a biochemical abnormality, but no manifestation of adrenal or neurologic disease. The cerebellum and brain stem are usually involved in individuals with olivo-ponto-cerebellar ALD, who present with signs between adolescence and adulthood.
The unique phenotype of the two brothers initially suggested a novel syndrome, rather than ALD. However, a missense mutation in ABCD1 was unexpectedly identified in both brothers and their mother by exome sequencing. Although PolyPhen-2 predicted that the biophysical consequences of the variant c.1126G > C (E376Q) of ABCD1 were likely to be functionally benign, other variants in the same exon, such as c.1114A > T (p.K372*) and c.1137C > G (p.S379R), have been reported to lead to ALD [15]. Further laboratory tests showed elevated plasma VLCFA levels in the brothers, and no mutations were detected in genes associated with other peroxisomal diseases, such as Zellweger syndrome, acyl-CoA oxidase deficiency, D-bi-functional protein deficiency, and b-ketothiolase deficiency. The brothers should be therefore diagnosed with ALD.
Nevertheless, the phenotype exhibited by the two brothers was not consistent with any reported ALD forms. Their ages at onset would suggest childhood cerebral ALD (CCALD), but their conditions were not progressive, their brain MRI results were normal, and they had severe congenital language and motor disorders and intellectual disability. In contrast, cognitive and motor development are normal in CCALD prior to the onset of demyelinating lesions visible at brain MRI, suggesting that the brothers' phenotype differed from CCALD. In addition, both brothers had urinary and fecal incontinence, their ages at onset were younger than 2 years old, they had severe congenital intellectual disabilities, and were unable to walk and speak, indicating a phenotype incompatible with AMN and other known forms of ALD. The distinct phenotype displayed by the two brothers suggests that other pathogenetic factors, in addition to ABCD1 mutation, may have been responsible for their condition.
DACH2 is a homolog of dachshund. The dachshund/Dach gene family encodes transcriptional cofactors that are conserved between insects and vertebrate. Drosophila dachshund is a critical regulator of eye, brain, and limb formation, and null mutations in dachshund result in abnormal retinal, brain, genital, and limb development [16]-[19]. The vertebrate homologs Dach1 and Dach2 also play an important role in the development of the retina, brain and limbs [20]. DACH2 encodes a transcription factor characterized by the presence of three conserved domains [21], which is involved in the regulation of organogenesis, myogenesis, brain and limb development [20],[22]. The first domain (DD1) at the N-terminus (amino acids 66-162) and the second domain (DD2) at the C-terminus (amino acids 452-543) are highly conserved in all members of the DACH protein family and appear to be involved in DNA binding and in the interaction with EYA proteins, respectively [23],[24]. A third domain DD3 is present in the central portion of the protein (amino acids 314-412) and is shared by all members of the DACH1 and DACH2 subfamilies, but its detailed function remains unknown. The variant c.1069A > T transition (p.S357C) of DACH2 detected in the present study was located in the DD3 domain and was predicted to be functionally damaging by PolyPhen-2, suggesting a potential role in two brothers- phenotype. The combined mutations in ABCD1 and DACH2 thus provide a plausible explanation for the abnormal phenotype observed in both brothers. However, further functional studies are needed to clarify the effects of these variants.
Conclusion
The distinct phenotype demonstrated by two brothers might represent a new form of ALD or a new syndrome. The combination of mutations in ABCD1 and DACH2 provides a plausible mechanism for this phenotype.
References
Moser HW, Raymond GV, Dubey P: Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA. 2005, 294 (24): 3131-3134. 10.1001/jama.294.24.3131.
Cappa M, Bizzarri C, Vollono C, Petroni A, Banni S: Adrenoleukodystrophy. Endocr Dev. 2011, 20: 149-160. 10.1159/000321236.
Powers JM: Adreno-leukodystrophy (adreno-testiculo-leukomyelo-neuropathic-complex). Clin Neuropathol. 1985, 4 (5): 181-199.
Powers JM, Liu Y, Moser AB, Moser HW: The inflammatory myelinopathy of adreno-leukodystrophy: cells, effector molecules, and pathogenetic implications. J Neuropathol Exp Neurol. 1992, 51 (6): 630-643. 10.1097/00005072-199211000-00007.
Fogel BL, Young P, Thompson AR, Perlman S: A family with combined mutations of the hemophilia A and X-linked adrenoleukodystrophy genes. Neurogenetics. 2008, 9 (3): 215-218. 10.1007/s10048-008-0132-6.
Corzo D, Gibson W, Johnson K, Mitchell G, LePage G, Cox GF, Casey R, Zeiss C, Tyson H, Cutting GR, Raymond GV, Smith KD, Watkins PA, Moser AB, Moser HW, Steinberg SJ: Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet. 2002, 70 (6): 1520-1531. 10.1086/340849.
Zhang Y, Dai Y, Liu Y, Ren J: Mandibulofacial dysostosis, microtia, and limb anomalies in a newborn: a new form of acrofacial dysostosis syndrome?. Clin Genet. 2010, 78 (6): 570-574. 10.1111/j.1399-0004.2010.01427.x.
Moser HW: Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997, 120 (Pt 8): 1485-1508. 10.1093/brain/120.8.1485.
van den Hurk JA, Schwartz M, van Bokhoven H, van de Pol TJ, Bogerd L, Pinckers AJ, Bleeker-Wagemakers EM, Pawlowitzki IH, Ruther K, Ropers HH, Cremers FP: Molecular basis of choroideremia (CHM): mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat. 1997, 9 (2): 110-117. 10.1002/(SICI)1098-1004(1997)9:2<110::AID-HUMU2>3.0.CO;2-D.
Takano H, Koike R, Onodera O, Sasaki R, Tsuji S: Mutational analysis and genotype-phenotype correlation of 29 unrelated Japanese patients with X-linked adrenoleukodystrophy. Arch Neurol. 1999, 56 (3): 295-300. 10.1001/archneur.56.3.295.
Kemp S, Pujol A, Waterham HR, van Geel BM, Boehm CD, Raymond GV, Cutting GR, Wanders RJ, Moser HW: ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum Mutat. 2001, 18 (6): 499-515. 10.1002/humu.1227.
Cartier N, Aubourg P: Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linked adrenoleukodystrophy. Brain Pathol. 2010, 20 (4): 857-862. 10.1111/j.1750-3639.2010.00394.x.
Moser HW, Mahmood A, Raymond GV: X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007, 3 (3): 140-151. 10.1038/ncpneuro0421.
Soardi FC, Esquiaveto-Aun AM, Guerra-Junior G, Lemos-Marini SH, Mello MP: Phenotypic variability in a family with x-linked adrenoleukodystrophy caused by the p.Trp132Ter mutation. Arq Bras Endocrinol Metabol. 2010, 54 (8): 738-743. 10.1590/S0004-27302010000800013.
X-linked adrenoleukodystrophy database. ., [http://www.x-ald.nl/]
Mardon G, Solomon NM, Rubin GM: dachshund encodes a nuclear protein required for normal eye and leg development in Drosophila. Development. 1994, 120 (12): 3473-3486.
Martini SR, Roman G, Meuser S, Mardon G, Davis RL: The retinal determination gene, dachshund, is required for mushroom body cell differentiation. Development. 2000, 127 (12): 2663-2672.
Keisman EL, Christiansen AE, Baker BS: The sex determination gene doublesex regulates the A/P organizer to direct sex-specific patterns of growth in the Drosophila genital imaginal disc. Dev Cell. 2001, 1 (2): 215-225. 10.1016/S1534-5807(01)00027-2.
Miguel-Aliaga I, Allan DW, Thor S: Independent roles of the dachshund and eyes absent genes in BMP signaling, axon pathfinding and neuronal specification. Development. 2004, 131 (23): 5837-5848. 10.1242/dev.01447.
Davis RJ, Shen W, Heanue TA, Mardon G: Mouse Dach, a homologue of Drosophila dachshund, is expressed in the developing retina, brain and limbs. Dev Genes Evol. 1999, 209 (9): 526-536. 10.1007/s004270050285.
Bione S, Rizzolio F, Sala C, Ricotti R, Goegan M, Manzini MC, Battaglia R, Marozzi A, Vegetti W, Dalprà L, Crosignani PG, Ginelli E, Nappi R, Bernabini S, Bruni V, Torricelli F, Zuffardi O, Toniolo D: Mutation analysis of two candidate genes for premature ovarian failure, DACH2 and POF1B. Hum Reprod. 2004, 19 (12): 2759-2766. 10.1093/humrep/deh502.
Ayres JA, Shum L, Akarsu AN, Dashner R, Takahashi K, Ikura T, Slavkin HC, Nuckolls GH: DACH: genomic characterization, evaluation as a candidate for postaxial polydactyly type A2, and developmental expression pattern of the mouse homologue. Genomics. 2001, 77 (1-2): 18-26. 10.1006/geno.2001.6618.
Davis RJ, Shen W, Sandler YI, Heanue TA, Mardon G: Characterization of mouse Dach2, a homologue of Drosophila dachshund. Mech Dev. 2001, 102 (1-2): 169-179. 10.1016/S0925-4773(01)00307-0.
Kim SS, Zhang RG, Braunstein SE, Joachimiak A, Cvekl A, Hegde RS: Structure of the retinal determination protein Dachshund reveals a DNA binding motif. Structure (Camb). 2002, 10 (6): 787-795. 10.1016/S0969-2126(02)00769-4.
Acknowledgments
We gratefully acknowledge the participation of all the participants.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YZ designed and led this study, analyzed test results and wrote the manuscript. YL, KZ and JW initiated the laboratory investigations of the patients, collected samples and provided the phenotype information. YL and YD(Yong Duan) confirmed and analyzed test results and reviewed the manuscript. YD(Yong Dai) was responsible for this study and reviewed the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhang, Y., Liu, Y., Li, Y. et al. Exome sequencing identifies mutations in ABCD1 and DACH2in two brothers with a distinct phenotype. BMC Med Genet 15, 105 (2014). https://doi.org/10.1186/s12881-014-0105-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-014-0105-6